A New Neurological Syndrome with Mental Retardation
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

General Contribution
24 Abstracts of 37th Annual Meeting A1 A SCREENING METHOD FOR FRAGILE X MUTATION: DETECTION OF THE CGG REPEAT IN FMR-1 GENE BY PCR WITH BIOTIN-LABELED PRIMER. ..Eiji NANBA, Kousaku OHNO and Kenzo TAKESHITA Division of Child Neurology, Institute of Neurological Sciences, Tot- tori University School of Medicine. Yonago We have developed a new polymerase chain reaction(PCR)-based method for detection of the CGG repeat in FMR-1 gene. No specific product from PCR was detected on the gel with ethidium bromide staining, because 7-deaza-2'-dGTP is necessary for amplification of this repeat. Biotin-labeled primer was used for PCR and the product was transferred to a nylon membrane followed the detection of biotin by Smilight kit. The size of PCR product from normal control were slightly various and around 300bp. No PCR product was detected from 3 fragile X male patients in 2 families diagnosed by cytogenetic examination. This method is useful for genetic screen- ing of male mental retardation patients to exclude the fragile X mutation. A2 DNA ANALYSISFOR FRAGILE X SYNDROME Osamu KOSUDA,Utak00GASA, ~.ideynki INH, a~ji K/NAGIJCltI, and Kazumasa ]tIKIJI (SILL Inc., Tokyo) Fragile X syndrome is X-linked disease having the amplification of (CG6)n repeat sequence in the chromsomeXq27.3. We performed Southern blot analysis using three probes recognized repetitive sequence resion. Normal controle showed 5.2Kb with Eco RI digest and 2.7Kb with Eco RI/Bss ttII digest as the germ tines by the Southern blot analysis. However, three cell lines established fro~ unrelated the patients with fragile X showed the abnormal bands between 5.2 and 7.7Kb with Eco RI digest, and between 2.7 and 7.7Kb with Eco aI/Bss HII digest. -

SUPPLEMENTARY MATERIAL Effect of Next
SUPPLEMENTARY MATERIAL Effect of Next-Generation Exome Sequencing Depth for Discovery of Diagnostic Variants KKyung Kim1,2,3†, Moon-Woo Seong4†, Won-Hyong Chung3, Sung Sup Park4, Sangseob Leem1, Won Park5,6, Jihyun Kim1,2, KiYoung Lee1,2*‡, Rae Woong Park1,2* and Namshin Kim5,6** 1Department of Biomedical Informatics, Ajou University School of Medicine, Suwon 443-749, Korea 2Department of Biomedical Science, Graduate School, Ajou University, Suwon 443-749, Korea, 3Korean Bioinformation Center, Korea Research Institute of Bioscience and Biotechnology, Daejeon 305-806, Korea, 4Department of Laboratory Medicine, Seoul National University Hospital College of Medicine, Seoul 110-799, Korea, 5Department of Functional Genomics, Korea University of Science and Technology, Daejeon 305-806, Korea, 6Epigenomics Research Center, Genome Institute, Korea Research Institute of Bioscience and Biotechnology, Daejeon 305-806, Korea http//www. genominfo.org/src/sm/gni-13-31-s001.pdf Supplementary Table 1. List of diagnostic genes Gene Symbol Description Associated diseases ABCB11 ATP-binding cassette, sub-family B (MDR/TAP), member 11 Intrahepatic cholestasis ABCD1 ATP-binding cassette, sub-family D (ALD), member 1 Adrenoleukodystrophy ACVR1 Activin A receptor, type I Fibrodysplasia ossificans progressiva AGL Amylo-alpha-1, 6-glucosidase, 4-alpha-glucanotransferase Glycogen storage disease ALB Albumin Analbuminaemia APC Adenomatous polyposis coli Adenomatous polyposis coli APOE Apolipoprotein E Apolipoprotein E deficiency AR Androgen receptor Androgen insensitivity -

Opthalmic Genetics
Inherited Genetics Opthalmic Genetics What is Opthalmic Genetics? Developmental • Anterior segment dysgenesis Of the approximately 5000 genetic diseases and syndromes known to affect humans, at • Aniridia least one-third involve the eye. Due to advances in molecular genetics and sequencing • Anophthlamos/ microphthalmos/ nanophthalmos methods, there has been an exponential increase in the knowledge of genetic eye diseases and syndromes. • Coloboma Complex Prevalence • Cataract • Keratoconus • More than 60% of cases of blindness among infants are caused by inherited eye • Fuchs endothelial corneal dystrophy diseases such as congenital cataracts, congenital glaucoma, retinal degeneration, • Age-related macular degeneration optic atrophy, eye malformations and corneal dystrophies. • Glaucoma • Pseudoexfoliation syndrome What are the common Genetic Opthalmic • Diabetic retinopathy Disorders ? Why do you need to test for Genetic Ophthalmic Disorders can be classified according to the type of genetic abnormality Opthalmic Disorders? Monogenic • There is also evidence now that the most common vision problems among children and adults are genetically determined (Eg: strabismus, amblyopia, refractive errors • Corneal dystrophies such as myopia, hyperopia and astigmatism) • Oculocutaneous Albinism • Genetic ophthalmic disorders include a large number of ocular pathologies which • Norrie disease have autosomal dominant, autosomal recessive or X-linked inheritance patterns, or • Retinoschisis are complex traits with polygenic and environmental components -

Genetic Investigation of 211 Chinese Families Expands the Mutational and Phenotypical Spectrum in Hereditary Retinopathy Genes Through Targeted Sequencing Technology
Genetic investigation of 211 Chinese families expands the mutational and phenotypical spectrum in hereditary retinopathy genes through targeted sequencing technology Zhouxian Bai The First Aliated Hospital of Zhengzhou University https://orcid.org/0000-0001-7071-666X Yanchuan Xie First Aliated Hospital of Henan University of Science and technology Lina Liu First Aliated Hospital of Zhengzhou University Jingzhi Shao Southern Medical University Nanfang Hospital Yuying Liu First Aliated Hospital of Zhengzhou University Xiangdong Kong ( [email protected] ) https://orcid.org/0000-0003-0030-7638 Research article Keywords: hereditary retinopathy, novel mutations, targeted sequencing, genetic testing Posted Date: December 4th, 2020 DOI: https://doi.org/10.21203/rs.3.rs-20958/v2 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Version of Record: A version of this preprint was published at BMC Medical Genomics on March 29th, 2021. See the published version at https://doi.org/10.1186/s12920-021-00935-w. Page 1/22 Abstract Background: Hereditary retinopathy is a signicant cause of blindness worldwide. Despite the discovery of many mutations in various retinopathies, a large part of patients remain undiagnosed genetically. Targeted next generation sequencing of the human genome is a suitable approach for retinopathy molecular diagnosis. Methods: We described a cohort of 211 families from central China with various forms of retinopathy, 95 families of which were investigated using NGS multi-gene panel sequencing as well as the other 116 patients were LHON suspected tested by Sanger sequencing. We validated the candidate variants by PCR- based Sanger sequencing. We have made comprehensive analysis of the cases through sequencing data and ophthalmologic examination information. -

Neurology, Neuromuscular, and Cardiology Disorders
GENETIC TESTING SOLUTIONS FOR: NEUROLOGY, NEUROMUSCULAR, AND CARDIOLOGY DISORDERS EGL Genetics has nearly 50 years of genetic testing history built upon a strong academic foundation. Our expertise spans common and rare genetic disease testing, genomic variant interpretation, test development and research. As we have grown, we have evolved into a high-science and high-performing CLIA-certifi ed and CAP-accredited laboratory with over 1,100 test offerings across biochemical genetics, cytogenetics, and molecular genetic testing. COMPREHENSIVE OFFERINGS There are signifi cant genetic and phenotypic heterogeneity for disorders involving neurology, neuromuscular, and cardiovascular systems, and obtaining a specifi c diagnosis is important for prognosis, patient management, and development of therapeutic strategies. Consider this collection of test offerings for patients with heart disease, intellectual disability, autism spectrum disorders, movement disorders, epilepsy, and more. Identifi cation of a causative genetic defect may provide information for prognosis and therapeutic intervention, and is required for carrier testing and early prenatal diagnosis. TEST OFFERINGS FOR THE FOLLOWING CLINICAL INDICATIONS: • Cardiomyopathy and cardiovascular diseases • Autism and intellectual disabilities • Neuromuscular disorders and muscular dystrophy • Epilepsy • Movement disorders ADVANTAGES OF PARTNERING WITH EGL GENETICS: • Board-certifi ed laboratory directors & genetic counselors to answer clinical and analytical questions • Multiple sample collection -

Pre-Implantation Genetic Diagnosis and Assisted Reproductive Technology in Haemophilia
PRE-IMPLANTATION GENETIC DIAGNOSIS AND ASSISTED REPRODUCTIVE TECHNOLOGY IN HAEMOPHILIA DR PENELOPE FOSTER MELBOURNE IVF WHAT IS PGD ? early embryo diagnosis allows selection of unaffected embryos for transfer to patient alternative to antenatal testing and termination of affected pregnancy MELBOURNE IVF TECHNIQUE OF PGD standard IVF cycle biopsy of 1 or 2 cells from day 3 embryo diagnostic testing on biopsied cells selection of embryos for transfer MELBOURNE IVF PGD IN HAEMOPHILIA OPTIONS Sex selection: if affected husband, all male offspring unaffected, all females carriers = select male embryos for transfer if carrier wife, 1/2 males affected, 1/2 females carrier = select female embryos for transfer MELBOURNE IVF PGD IN HAEMOPHILIA OPTIONS Specific gene detection avoids discarding unaffected male embryos avoids transfer of carrier female embryos MELBOURNE IVF SEX SELECTION - FISH FLUORESCENT IN-SITU HYBRIDISATION detects chromosome number cell from embryo fixed to slide apply FISH probes DNA sequences complementary to small segment of particular chromosome probes labelled with coloured fluorochromes coloured spots indicate presence of sequence 8-probe FISH – chromosomes 4,13,16,18,21,22,X,Y select euploid XX or XY embryos for transfer MELBOURNE IVF FISH ON BLASTOMERES 13, 16, 18, 21, 22 X, Y, 4 MELBOURNE IVF XX XY MELBOURNE IVF PGD FOR SPECIFIC GENE DETECTION DNA amplification by PCR 2 cells from embryo fragment analysis on DNA sequencer inclusion of informative markers individualised tests for each couple significant -

Manifesting Heterozygosity in Norrie's Disease?
British3Journal ofOphthalmology 1993; 1i3813 CASE REPORTS Manifesting heterozygosity in Norrie's disease? G Woodruff, R Newbury-Ecob, D S Plaha, I D Young It is well recognised that Norrie's disease is an ii-2 X linked disorder causing blindness in early This 30-year-old man is of normal intelligence. infancy, often in association with hearing loss Assessment at age 22 years showed bilateral and/or psychomotor retardation.' The diagnosis pthisical eyes. is established by the finding of congenital pseudoglioma in a male infant with either typical systemic features or a family history of congeni- II-4 Department of Ophthalmology, tal blindness in male relatives.'2 We report here The 32-year-old mother of the proband (III-1) is University ofLeicester the ocular findings in a 2-year-old girl who of above average intelligence and has normal G Woodruff appears to be a manifesting carrier of Norrie's vision. Funduscopy and fluorescein angiography Department ofClinical disease, this being an observation which has were normal. Genetics, City Hospital, important implications for genetic counselling. Nottingham R Newbury-Ecob I D Young Case reports This 8-year-old boy was found to have bilateral Molecular Genetics The family pedigree is shown in Figure 1. total retinal detachment with retrolental masses Laboratory, Leicester at age 2 weeks. Royal Infirmary, and absent electroretinogram Leicester Horizontal corneal diameters were 10 mm (right) D S Plaha II-1 and 9 mm (left). Initial development appeared Correspondence to: This deceased male lived in Mauritius and was normal but by 3 years marked global delay was Mr G Woodruff, Department ofOphthalmology, Clinical first noted to be blind at age 2 years. -

Norrie Disease Resulting from a Gene Deletion: Clinical Features and DNA Studies
J Med Genet: first published as 10.1136/jmg.25.2.73 on 1 February 1988. Downloaded from Journal of Medical Genetics 1988, 25, 73-78 Norrie disease resulting from a gene deletion: clinical features and DNA studies DIAN DONNAI, R C MOUNTFORD, AND A P READ From the Department of Medical Genetics, St Mary's Hospital, Manchester M13 OJH. SUMMARY We describe a family in which two boys with Norrie disease have a deletion of the DXS7 locus. The deletion does not extend as far distally as the OTC or DXS84 loci. A full clinical description of the patients is given to help establish the range of manifestations of Norrie disease. There is no evidence of any other X linked disease in our patients. Norrie disease (NDP, McKusick No 31060) was guineous parents after a normal pregnancy. Deli-'- originally described as X linked blindness.' It is very was normal and his birth weight was 2-6 kg at characterised by early vascular proliferation term. His mother noted that his eyes looked cloudy (pseudoglioma) in both retinas, atrophic irides, on the eighth day of life. After ophthalmic evalua- corneal clouding, and cataracts, progressing to tion a diagnosis of primary hyperplastic vitreous was shrinkage of the globes (phthisis bulbi). The eye suggested and his left eye was enucleated at 14 findings are well described,25 but extraocular months because of shrinkage, pain, and the possi- abnormalities are less commonly illustrated. Diag- bility of a tumour. Developmental delay was ap- copyright. nosis is usually made on ophthalmological criteria, parent from six months but investigations at that but we believe that in many cases the disorder is time did not identify the cause. -
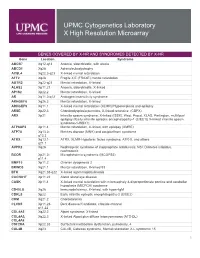
Genes Covered and Disorders Detected by X-HR Microarray
UPMC Cytogenetics Laboratory X High Resolution Microarray GENES COVERED BY X-HR AND SYNDROMES DETECTED BY X-HR Gene Location Syndrome ABCB7 Xq12-q13 Anemia, sideroblastic, with ataxia ABCD1 Xq28 Adrenoleukodystrophy ACSL4 Xq22.3-q23 X-linked mental retardation AFF2 Xq28 Fragile X E (FRAXE) mental retardation AGTR2 Xq22-q23 Mental retardation, X-linked ALAS2 Xp11.21 Anemia, sideroblastic, X-linked AP1S2 Xp22.2 Mental retardation, X-linked AR Xq11.2-q12 Androgen insensitivity syndrome ARHGEF6 Xq26.3 Mental retardation, X-linked ARHGEF9 Xq11.1 X-linked mental retardation (XLMR)//Hyperekplexia and epilepsy ARSE Xp22.3 Chondrodysplasia punctata, X-linked recessive (CDPX) ARX Xp21 Infantile spasm syndrome, X-linked (ISSX), West, Proud, XLAG, Partington, multifocal epilepsy //Early infantile epileptic encephalopathy-1 (EIEE1)( X-linked infantile spasm syndrome-1-ISSX1) ATP6AP2 Xp11.4 Mental retardation, X-linked, with epilepsy (XMRE) ATP7A Xq13.2- Menkes disease (MNK) and occipital horn syndrome q13.3 ATRX Xq13.1- ATRX, XLMR-Hypotonic facies syndrome, ATR-X, and others q21.1 AVPR2 Xq28 Nephrogenic syndrome of inappropriate antidiuresis; NSI; Diabetes insipidus, nephrogenic BCOR Xp21.2- Microphthalmia syndromic (MCOPS2) p11.4 BMP15 Xp11.2 Ovarian dysgenesis 2 BRWD3 Xq21.1 Mental retardation, X-linked 93 BTK Xq21.33-q22 X-linked agammaglobulinemia CACNA1F Xp11.23 Aland Island eye disease CASK Xp11.4 X-linked mental retardation with microcephaly & disproportionate pontine and cerebellar hypoplasia (MICPCH) syndrome CD40LG Xq26 Immunodeficiency, -

Norrie Disease
Norrie disease Description Norrie disease is an inherited eye disorder that leads to blindness in male infants at birth or soon after birth. It causes abnormal development of the retina, the layer of sensory cells that detect light and color, with masses of immature retinal cells accumulating at the back of the eye. As a result, the pupils appear white when light is shone on them, a sign called leukocoria. The irises (colored portions of the eyes) or the entire eyeballs may shrink and deteriorate during the first months of life, and cataracts ( cloudiness in the lens of the eye) may eventually develop. About 30 percent of individuals with Norrie disease develop progressive hearing loss, and 30 to 50 percent of people affected experience developmental delays in motor skills such as sitting up and walking. Other problems may include mild to moderate intellectual disability, often with psychosis, and abnormalities that can affect circulation, breathing, digestion, excretion, or reproduction. Frequency Norrie disease is a rare disorder; more than 400 cases have been reported in the scientific literature. Causes Mutations in the NDP gene cause Norrie disease. The NDP gene provides instructions for making a protein called norrin. Norrin participates in the Wnt cascade, a sequence of steps that affect the way cells and tissues develop. In particular, norrin seems to play a critical role in the specialization of retinal cells for their unique sensory capabilities. It is also involved in the establishment of a blood supply to tissues of the retina and the inner ear, and the development of other body systems. -

A Fetus with an X;1 Balanced Reciprocal Translocation and Eye
I Med Genet 1995;32:557-560 557 A fetus with an X; 1 balanced reciprocal translocation and eye disease J Med Genet: first published as 10.1136/jmg.32.7.557 on 1 July 1995. Downloaded from Mary J Seller, Kalyani Pal, Sharon Horsley, Angela F Davies, A Caroline Berry, R Meredith, Alison C E McCartney Abstract changes consistent with the early stages ofNor- A 19 week female fetus is described with a rie disease. de novo X;1 reciprocal balanced trans- location, with the breakpoint on the X chromosome at Xpll.4, and eye pathology Case report consistent with the early stages of Norrie This was the sixth pregnancy of a healthy white disease. The fetus seems to be an example couple aged 37 and 41 years. The previous of a female manifesting an X linked re- children, five boys and one girl, are well, in- cessive disease, and it was shown that the cluding a boy, one of twins and now 13 years normal X chromosome was completely old, who had surgery during infancy for trans- inactivated in all cells examined. Norrie position ofthe great vessels. The family history disease has been mapped to Xpll.3, and did not indicate anything significant except for fluorescence in situ hybridisation studies a niece on the paternal side who had multiple showed that the Norrie disease gene had abnormalities and died. No precise diagnosis not obviously been disrupted. Mutation had been made but a chromosome abnormality screening by SSCP analysis showed no was suspected. For this reason, and on grounds aberrant fragments of the coding region of maternal age, chorionic villus sampling was ofthe gene. -

Hereditary Hearing Loss and It's Syndromes
Syndromes and Hearing Loss – Clinical Practice Guideline for Audiology (this is a section of a larger Practice Guideline “Cleft Palate, Craniofacial and Syndromic Guideline”) Care Paths for these syndromes are in separate PDF files in the same place as this document was found. There are many known syndromes associated with hearing loss. Many of these have clefting and/or craniofacial anomalies, some of them don’t. This list was generated by combining the BCCH Audiology Department list of syndromes and the BCEHP Late Onset Monitoring Risk Factor Syndromes. That list was then compared with those found in the “Hereditary Hearing Loss and It’s Syndromes” and reviewed by all of the reviewers of this Guideline for completeness. This resulted in the syndromes listed below which are associated with hearing loss. A literature review was conducted using Pub Med, PEDLYNX, and OMIM databases. Search terms were (‘name of syndrome’ as listed in Appendix B AND (‘Audio*’ OR Hear*’) in title or abstract, from 1999 to 2010, all languages. Citations were screened by a two reviewers for relevance. Published, peer reviewed articles were selected based on level of evidence with recently published articles describing well-designed randomised controlled trials with comparatively large sample groups taking precedence. High quality systematic reviews and retrospective reviews of clinical data were also used. Case studies of noteworthy results were occasionally noted as a matter of interest or possible focus of higher level literature to be reviewed in the future (when published), but were not considered in determining association of a syndrome with late onset SNHL. If the results of a study were inconclusive or the literature could not clearly associate a syndrome with late onset SNHL (ie.