Role of FBXW5-Loss in Centrosome Abnormalities and Cell Physiology
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

PRODUCT SPECIFICATION Product Datasheet
Product Datasheet QPrEST PRODUCT SPECIFICATION Product Name QPrEST FBXW5 Mass Spectrometry Protein Standard Product Number QPrEST33470 Protein Name F-box/WD repeat-containing protein 5 Uniprot ID Q969U6 Gene FBXW5 Product Description Stable isotope-labeled standard for absolute protein quantification of F-box/WD repeat- containing protein 5. Lys (13C and 15N) and Arg (13C and 15N) metabolically labeled recombinant human protein fragment. Application Absolute protein quantification using mass spectrometry Sequence (excluding PDNRYLYVNSRAWPNGAVVADPMQPPPIAEEIDLLVFDLKTMREVRRALR fusion tag) AHRAYTPNDECFFIFLDVSRDFVASGAEDRHGYIWDRHYNICLARLRHED VVNSVVFSPQEQ Theoretical MW 30962 Da including N-terminal His6ABP fusion tag Fusion Tag A purification and quantification tag (QTag) consisting of a hexahistidine sequence followed by an Albumin Binding Protein (ABP) domain derived from Streptococcal Protein G. Expression Host Escherichia coli LysA ArgA BL21(DE3) Purification IMAC purification Purity >90% as determined by Bioanalyzer Protein 230 Purity Assay Isotopic Incorporation >99% Concentration >5 μM after reconstitution in 100 μl H20 Concentration Concentration determined by LC-MS/MS using a highly pure amino acid analyzed internal Determination reference (QTag), CV ≤10%. Amount >0.5 nmol per vial, two vials supplied. Formulation Lyophilized in 100 mM Tris-HCl 5% Trehalose, pH 8.0 Instructions for Spin vial before opening. Add 100 μL ultrapure H2O to the vial. Vortex thoroughly and spin Reconstitution down. For further dilution, see Application Protocol. Shipping Shipped at ambient temperature Storage Lyophilized product shall be stored at -20°C. See COA for expiry date. Reconstituted product can be stored at -20°C for up to 4 weeks. Avoid repeated freeze-thaw cycles. Notes For research use only Product of Sweden. For research use only. -

The ULK1-FBXW5-SEC23B Nexus Controls Autophagy
bioRxiv preprint doi: https://doi.org/10.1101/441923; this version posted October 12, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. The ULK1‐FBXW5‐SEC23B nexus controls autophagy Yeon‐Tae Jeong1,2, Daniele Simoneschi1,2, Sarah Keegan1,2,3, David Melville4, Natalia S. Adler5, Anita Saraf6, Laurence Florens6, Michael P. Washburn6,7, Claudio N. Cavasotto5, David Fenyö1,2,3, Ana‐Maria Cuervo8, Mario Rossi1,2,5, and Michele Pagano1,2,9* 1Department of Biochemistry and Molecular Pharmacology, 2Perlmutter NYU Cancer Center, 3Institute for System Genetics, and 9Howard Hughes Medical Institute; New York University School of Medicine, 522 First Avenue, New York, NY 10016, USA. 4Department of Molecular and Cellular Biology and Howard Hughes Medical Institute, University of California, Berkeley, CA, USA. 5Instituto de Investigación en Biomedicina de Buenos Aires‐ CONICET‐‐Partner Institute of the Max Planck Society, C1425FQD Buenos Aires, Argentina. 6The Stowers Institute for Medical Research, 1000 East 50th Street, Kansas City, MO 64110, USA. 7Department of Pathology and Laboratory Medicine, The University of Kansas Medical Center, 3901 Rainbow Boulevard, Kansas City, Kansas 66160, USA. 8Department of Developmental and Molecular Biology and Institute for Aging Studies, Albert Einstein College of Medicine, Bronx, NY USA. *Correspondence: [email protected] 1 | Page bioRxiv preprint doi: https://doi.org/10.1101/441923; this version posted October 12, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. -

UC Berkeley UC Berkeley Electronic Theses and Dissertations
UC Berkeley UC Berkeley Electronic Theses and Dissertations Title Networks of Splice Factor Regulation by Unproductive Splicing Coupled With NMD Permalink https://escholarship.org/uc/item/4md923q7 Author Desai, Anna Publication Date 2017 Peer reviewed|Thesis/dissertation eScholarship.org Powered by the California Digital Library University of California Networks of Splice Factor Regulation by Unproductive Splicing Coupled With NMD by Anna Maria Desai A dissertation submitted in partial satisfaction of the requirements for the degree of Doctor of Philosophy in Comparative Biochemistry in the Graduate Division of the University of California, Berkeley Committee in charge: Professor Steven E. Brenner, Chair Professor Donald Rio Professor Lin He Fall 2017 Abstract Networks of Splice Factor Regulation by Unproductive Splicing Coupled With NMD by Anna Maria Desai Doctor of Philosophy in Comparative Biochemistry University of California, Berkeley Professor Steven E. Brenner, Chair Virtually all multi-exon genes undergo alternative splicing (AS) to generate multiple protein isoforms. Alternative splicing is regulated by splicing factors, such as the serine/arginine rich (SR) protein family and the heterogeneous nuclear ribonucleoproteins (hnRNPs). Splicing factors are essential and highly conserved. It has been shown that splicing factors modulate alternative splicing of their own transcripts and of transcripts encoding other splicing factors. However, the extent of this alternative splicing regulation has not yet been determined. I hypothesize that the splicing factor network extends to many SR and hnRNP proteins, and is regulated by alternative splicing coupled to the nonsense mediated mRNA decay (NMD) surveillance pathway. The NMD pathway has a role in preventing accumulation of erroneous transcripts with dominant negative phenotypes. -

Human Leucine-Rich Repeat Proteins: a Genome-Wide Bioinformatic Categorization and Functional Analysis in Innate Immunity
Human leucine-rich repeat proteins: a genome-wide bioinformatic categorization and functional analysis in innate immunity Aylwin C. Y. Nga,b,1, Jason M. Eisenberga,b,1, Robert J. W. Heatha, Alan Huetta, Cory M. Robinsonc, Gerard J. Nauc, and Ramnik J. Xaviera,b,2 aCenter for Computational and Integrative Biology, and Gastrointestinal Unit, Massachusetts General Hospital and Harvard Medical School, Boston, MA 02114; bThe Broad Institute of Massachusetts Institute of Technology and Harvard, Cambridge, MA 02142; and cMicrobiology and Molecular Genetics, University of Pittsburgh School of Medicine, Pittsburgh, PA 15261 Edited by Jeffrey I. Gordon, Washington University School of Medicine, St. Louis, MO, and approved June 11, 2010 (received for review February 17, 2010) In innate immune sensing, the detection of pathogen-associated proteins have been implicated in human diseases to date, notably molecular patterns by recognition receptors typically involve polymorphisms in NOD2 in Crohn disease (8, 9), CIITA in leucine-rich repeats (LRRs). We provide a categorization of 375 rheumatoid arthritis and multiple sclerosis (10), and TLR5 in human LRR-containing proteins, almost half of which lack other Legionnaire disease (11). identifiable functional domains. We clustered human LRR proteins Most LRR domains consist of a chain of between 2 and 45 by first assigning LRRs to LRR classes and then grouping the proteins LRRs (12). Each repeat in turn is typically 20 to 30 residues long based on these class assignments, revealing several of the resulting and can be divided into a highly conserved segment (HCS) fol- protein groups containing a large number of proteins with certain lowed by a variable segment (VS). -

A Novel 2.3 Mb Microduplication of 9Q34. 3 Inserted Into 19Q13. 4 in A
Hindawi Publishing Corporation Case Reports in Pediatrics Volume 2012, Article ID 459602, 7 pages doi:10.1155/2012/459602 Case Report A Novel 2.3 Mb Microduplication of 9q34.3 Inserted into 19q13.4 in a Patient with Learning Disabilities Shalinder Singh,1 Fern Ashton,1 Renate Marquis-Nicholson,1 Jennifer M. Love,1 Chuan-Ching Lan,1 Salim Aftimos,2 Alice M. George,1 and Donald R. Love1, 3 1 Diagnostic Genetics, LabPlus, Auckland City Hospital, P.O. Box 110031, Auckland 1148, New Zealand 2 Genetic Health Service New Zealand-Northern Hub, Auckland City Hospital, Private Bag 92024, Auckland 1142, New Zealand 3 School of Biological Sciences, University of Auckland, Private Bag 92019, Auckland 1142, New Zealand Correspondence should be addressed to Donald R. Love, [email protected] Received 1 July 2012; Accepted 27 September 2012 Academic Editors: L. Cvitanovic-Sojat, G. Singer, and V. C. Wong Copyright © 2012 Shalinder Singh et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Insertional translocations in which a duplicated region of one chromosome is inserted into another chromosome are very rare. We report a 16.5-year-old girl with a terminal duplication at 9q34.3 of paternal origin inserted into 19q13.4. Chromosomal analysis revealed the karyotype 46,XX,der(19)ins(19;9)(q13.4;q34.3q34.3)pat. Cytogenetic microarray analysis (CMA) identified a ∼2.3Mb duplication of 9q34.3 → qter, which was confirmed by Fluorescence in situ hybridisation (FISH). -

FBXW5 Polyclonal Antibody Catalog Number PA5-31635 Product Data Sheet
Lot Number: TG2603497D Website: thermofisher.com Customer Service (US): 1 800 955 6288 ext. 1 Technical Support (US): 1 800 955 6288 ext. 441 thermofisher.com/contactus FBXW5 Polyclonal Antibody Catalog Number PA5-31635 Product Data Sheet Details Species Reactivity Size 100 µL Tested species reactivity Human Host / Isotype Rabbit IgG Tested Applications Dilution * Class Polyclonal Immunohistochemistry (Paraffin) 1:100-1:1000 Type Antibody (IHC (P)) Recombinant fragment Western Blot (WB) 1:500-1:3000 corresponding to a region within Immunogen * Suggested working dilutions are given as a guide only. It is recommended that the user titrate the product for use in their amino acids 177 and 432 of Human own experiment using appropriate negative and positive controls. FBXW5 Conjugate Unconjugated Form Liquid Concentration 1mg/mL Purification Antigen affinity chromatography Storage Buffer 0.1M tris glycine, pH 7, with 20% glycerol Contains 0.01% thimerosal Storage Conditions -20° C, Avoid Freeze/Thaw Cycles Product Specific Information PA5-31635 targets FBXW5 in IHC (P) and WB applications and shows reactivity with Human samples. The PA5-31635 immunogen is recombinant fragment corresponding to a region within amino acids 177 and 432 of Human FBXW5. Background/Target Information This gene encodes a member of the F-box protein family, members of which are characterized by an approximately 40 amino acid motif, the F-box. The F-box proteins constitute one of the four subunits of ubiquitin protein ligase complex called SCFs (SKP1-cullin-F-box), which function in phosphorylation-dependent ubiquitination. The F-box proteins are divided into three classes: Fbws containing WD-40 domains, Fbls containing leucine-rich repeats, and Fbxs containing either different protein-protein interaction modules or no recognizable motifs. -
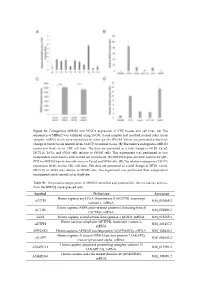
Figure S1. Endogenous MIR45
Figure S1. Endogenous MIR452 and VEGFA expression in CRC tissues and cell lines. (A) The expression of MIR452 was validated using 10 CRC tissue samples and matched normal colon tissue samples. miRNA levels were normalized to colon-specific RNU48. Values are presented as the fold- change in tumor tissue relative levels (ΔΔCT) to normal tissue. (B) The relative endogenous MIR452 expression levels in six CRC cell lines. The data are presented as a fold change in HT29, Caco2, HCT116, LoVo, and SW48 cells relative to SW480 cells. This experiment was performed as two independent experiments, each carried out in triplicate. (C) MIR452 expression level analysis by qRT- PCR for MIR452 transfection efficiency in Caco2 and SW48 cells. (D) The relative endogenous VEGFA expression levels in five CRC cell lines. The data are presented as a fold change in HT29, Caco2, HCT116, or SW48 cells relative to SW480 cells. This experiment was performed three independent experiments, each carried out in duplicate. Table S1. The putative target genes of MIR452 identified and predicted by the microarray analysis from the MIR452 overexpressed cells. Symbol Definition Accession Homo sapiens acyl-CoA thioesterase 8 (ACOT8), transcript ACOT8 NM_005469.2 variant 1, mRNA. Homo sapiens ARP6 actin-related protein 6 homolog (yeast) ACTR6 NM_022496.3 (ACTR6), mRNA. ADI1 Homo sapiens acireductone dioxygenase 1 (ADI1), mRNA. NM_018269.1 Homo sapiens aftiphilin (AFTPH), transcript variant 1, AFTPH NM_203437.2 mRNA. AHNAK2 Homo sapiens AHNAK nucleoprotein 2 (AHNAK2), mRNA. NM_138420.2 Homo sapiens A kinase (PRKA) anchor protein 7 (AKAP7), AKAP7 NM_004842.2 transcript variant alpha, mRNA. Homo sapiens anaphase promoting complex subunit 13 ANAPC13 NM_015391.2 (ANAPC13), mRNA. -

The ULK1-FBXW5-SEC23B Nexus Controls Autophagy
RESEARCH ARTICLE The ULK1-FBXW5-SEC23B nexus controls autophagy Yeon-Tae Jeong1,2, Daniele Simoneschi1,2, Sarah Keegan1,2,3, David Melville4, Natalia S Adler5,6, Anita Saraf7, Laurence Florens7, Michael P Washburn7,8, Claudio N Cavasotto5,6, David Fenyo¨ 1,2,3, Ana Maria Cuervo9, Mario Rossi1,5,2*, Michele Pagano1,2,10* 1Department of Biochemistry and Molecular Pharmacology, NYU School of Medicine, New York, United States; 2Perlmutter NYU Cancer Center, NYU School of Medicine, New York, United States; 3Institute for System Genetics, NYU School of Medicine, New York, United States; 4Department of Molecular and Cellular Biology, Howard Hughes Medical Institute, University of California, Berkeley, Berkeley, United States; 5Instituto de Investigacio´n en Biomedicina de Buenos Aires (IBioBA), CONICET-Partner Institute of the Max Planck Society, Buenos Aires, Argentina; 6Translational Medicine Research Institute (IIMT), CONICET, Facultad de Ciencias Biome´dicas and Facultad deIngenierı´a, Universidad Austral, Pilar-Derqui, Argentina; 7The Stowers Institute for Medical Research, Kansas, United States; 8Department of Pathology and Laboratory Medicine, The University of Kansas Medical Center, Kansas, United States; 9Department of Developmental and Molecular Biology, Institute for Aging Studies, Albert Einstein College of Medicine, Bronx, United States; 10Howard Hughes Medical Institute, New York University School of Medicine, New York, United States *For correspondence: Abstract In response to nutrient deprivation, the cell mobilizes an extensive amount of [email protected]. membrane to form and grow the autophagosome, allowing the progression of autophagy. By ar (MR); providing membranes and stimulating LC3 lipidation, COPII (Coat Protein Complex II) promotes [email protected] (MP) autophagosome biogenesis. -

The Functional Role of SEC23 in Vesicle Transportation, Autophagy and Cancer Jingchen Jing1, 2, Bo Wang1, 2, Peijun Liu1, 2
Int. J. Biol. Sci. 2019, Vol. 15 2419 Ivyspring International Publisher International Journal of Biological Sciences 2019; 15 (11): 2419-2426. doi: 10.7150/ijbs.37008 Review The Functional Role of SEC23 in Vesicle Transportation, Autophagy and Cancer Jingchen Jing1, 2, Bo Wang1, 2, Peijun Liu1, 2 1. Center for Translational Medicine, The First Affiliated Hospital, Xi'an Jiaotong University 2. The Key Laboratory for Tumor Precision Medicine of Shaanxi Province, The First Affiliated Hospital, Xi'an Jiaotong University, Xi'an, Shaanxi 710061, China Corresponding author: Dr. Peijun Liu, Center for Translational Medicine, The First Affiliated Hospital, Xi'an Jiaotong University, 277 Yanta West Road, Xi'an, Shaanxi 710061, China. E‑mail: [email protected] © The author (s). This is an open access article distributed under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0/). See http://ivyspring.com/terms for full terms and conditions. Received: 2019.05.26; Accepted: 2019.08.01; Published: 2019.09.07 Abstract SEC23, the core component of the coat protein complex II (COPII), functions to transport newly synthesized proteins and lipids from the endoplasmic reticulum (ER) to the Golgi apparatus in cells for secretion. SEC23 protein has two isoforms (SEC23A and SEC23B) and their aberrant expression and mutations were reported to cause human diseases and oncogenesis, whereas SEC23A and SEC23B may have the opposite activity in human cancer, for a reason that remains unclear. This review summarizes recent research in SEC23, COPII-vesicle transportation, autophagy, and cancer. Key words: SEC23, COPII-vesicle transportation, autophagy, cancer Introduction SEC23 is one of the components in the coat SEC23, COPII-vesicle transportation, autophagy, and protein complex II (COPII) and regulates the cancer development and progression (Fig. -

FBXW5 Polyclonal Antibody
Product Datasheet FBXW5 Polyclonal Antibody Catalog No: #31706 Package Size: #31706-1 50ul #31706-2 100ul Orders: [email protected] Support: [email protected] Description Product Name FBXW5 Polyclonal Antibody Host Species Rabbit Clonality Polyclonal Isotype IgG Purification Affinity purification Applications WB Species Reactivity Human Immunogen Description Recombinant fusion protein of human FBXW5 (NP_061871.1). Other Names FBXW5; Fbw5; F-box/WD repeat-containing protein 5 Accession No. Swiss-Prot#:Q969U6NCBI Gene ID:54461 Calculated MW 72kDa Formulation Avoid freeze / thaw cycles.|Buffer: PBS with 50% glycerol, pH7.4. Storage Store at -20°C Application Details WB 1:200 - 1:1000 Images Western blot analysis of extracts of A-549 cells, using FBXW5 at 1:1000 dilution. Background This gene encodes a member of the F-box protein family, members of which are characterized by an approximately 40 amino acid motif, the F-box. The F-box proteins constitute one of the four subunits of ubiquitin protein ligase complex called SCFs (SKP1-cullin-F-box), which function in phosphorylation-dependent ubiquitination. The F-box proteins are divided into three classes: Fbws containing WD-40 domains, Fbls containing leucine-rich repeats, and Fbxs containing either different protein-protein interaction modules or no recognizable motifs. The protein encoded by this gene contains WD-40 domains, in addition to an F-box motif, so it belongs to the Fbw class. Alternatively spliced transcript variants encoding distinct isoforms have been identified for this gene, however, they were found to be nonsense-mediated mRNA decay (NMD) candidates, hence not represented. Address: 8400 Baltimore Ave., Suite 302, College Park, MD 20740, USA http://www.sabbiotech.com 1 Note: This product is for in vitro research use only and is not intended for use in humans or animals. -

FBXW5 Polyclonal Antibody
PRODUCT DATA SHEET Bioworld Technology,Inc. FBXW5 polyclonal antibody Catalog: BS72964 Host: Rabbit Reactivity: Human BackGround: Purification&Purity: This gene encodes a member of the F-box protein family, The antibody was affinity-purified from rabbit antiserum members of which are characterized by an approximately by affinity-chromatography using epitope-specific im- 40 amino acid motif, the F-box. The F-box proteins con- munogen and the purity is > 95% (by SDS-PAGE). stitute one of the four subunits of ubiquitin protein ligase Applications: complex called SCFs (SKP1-cullin-F-box), which func- WB 1:200 - 1:1000 tion in phosphorylation-dependent ubiquitination. The Storage&Stability: F-box proteins are divided into three classes: Fbws con- Store at 4°C short term. Aliquot and store at -20°C long taining WD-40 domains, Fbls containing leucine-rich re- term. Avoid freeze-thaw cycles. peats, and Fbxs containing either different protein-protein Specificity: interaction modules or no recognizable motifs. The pro- FBXW5 polyclonal antibody detects endogenous levels tein encoded by this gene contains WD-40 domains, in of FBXW5 protein. addition to an F-box motif, so it belongs to the Fbw class. DATA: Alternatively spliced transcript variants encoding distinct isoforms have been identified for this gene, however, they were found to be nonsense-mediated mRNA decay (NMD) candidates, hence not represented. Product: Rabbit IgG, 1mg/ml in PBS with 0.02% sodium azide, 50% glycerol, pH7.2 Molecular Weight: Western blot analysis of extracts of A-549 cells, using FBXW5 anti- 72kDa body. Swiss-Prot: Note: Q969U6 For research use only, not for use in diagnostic procedure. -
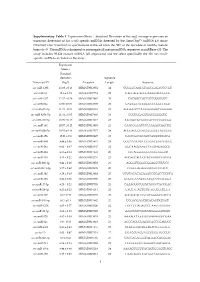
Supplementary Table 1. Expression
Supplementary Table 1. Expression (Mean Standard Deviation of the log2 average expression or transcript detection) of Sus scrofa specific miRNAs detected by the GeneChip™ miRNA 4.0 Array (ThermoFisher Scientific) in spermatozoa retrieved from the SRF of the ejaculate of healthy mature boars (n=3). The miRNA is designed to interrogate all mature miRNA sequences in miRBase v20. The array includes 30.424 mature miRNA (all organisms) and we select specifically the 326 Sus scrofa- specific miRNAs included in the array. Expression Mean ± Standard Deviation Sequence Transcript ID (log2) Accession Length Sequence ssc-miR-1285 13.98 ± 0.13 MIMAT0013954 24 CUGGGCAACAUAGCGAGACCCCGU ssc-miR-16 12.6 ± 0.74 MIMAT0007754 22 UAGCAGCACGUAAAUAUUGGCG ssc-miR-4332 12.32 ± 0.29 MIMAT0017962 20 CACGGCCGCCGCCGGGCGCC ssc-miR-92a 12.06 ± 0.09 MIMAT0013908 22 UAUUGCACUUGUCCCGGCCUGU ssc-miR-671-5p 11.73 ± 0.54 MIMAT0025381 24 AGGAAGCCCUGGAGGGGCUGGAGG ssc-miR-4334-5p 11.31 ± 0.05 MIMAT0017966 19 CCCUGGAGUGACGGGGGUG ssc-miR-425-5p 10.99 ± 0.15 MIMAT0013917 23 AAUGACACGAUCACUCCCGUUGA ssc-miR-191 10.57 ± 0.22 MIMAT0013876 23 CAACGGAAUCCCAAAAGCAGCUG ssc-miR-92b-5p 10.53 ± 0.18 MIMAT0017377 24 AGGGACGGGACGCGGUGCAGUGUU ssc-miR-15b 10.01 ± 0.9 MIMAT0002125 22 UAGCAGCACAUCAUGGUUUACA ssc-miR-30d 9.89 ± 0.36 MIMAT0013871 24 UGUAAACAUCCCCGACUGGAAGCU ssc-miR-26a 9.62 ± 0.47 MIMAT0002135 22 UUCAAGUAAUCCAGGAUAGGCU ssc-miR-484 9.55 ± 0.14 MIMAT0017974 20 CCCAGGGGGCGACCCAGGCU ssc-miR-103 9.53 ± 0.22 MIMAT0002154 23 AGCAGCAUUGUACAGGGCUAUGA ssc-miR-296-3p 9.41 ± 0.26 MIMAT0022958