Chromosome-Level Genome Assembly of a Butterflyfish, Chelmon Rostratus
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Pacific Plate Biogeography, with Special Reference to Shorefishes
Pacific Plate Biogeography, with Special Reference to Shorefishes VICTOR G. SPRINGER m SMITHSONIAN CONTRIBUTIONS TO ZOOLOGY • NUMBER 367 SERIES PUBLICATIONS OF THE SMITHSONIAN INSTITUTION Emphasis upon publication as a means of "diffusing knowledge" was expressed by the first Secretary of the Smithsonian. In his formal plan for the Institution, Joseph Henry outlined a program that included the following statement: "It is proposed to publish a series of reports, giving an account of the new discoveries in science, and of the changes made from year to year in all branches of knowledge." This theme of basic research has been adhered to through the years by thousands of titles issued in series publications under the Smithsonian imprint, commencing with Smithsonian Contributions to Knowledge in 1848 and continuing with the following active series: Smithsonian Contributions to Anthropology Smithsonian Contributions to Astrophysics Smithsonian Contributions to Botany Smithsonian Contributions to the Earth Sciences Smithsonian Contributions to the Marine Sciences Smithsonian Contributions to Paleobiology Smithsonian Contributions to Zoo/ogy Smithsonian Studies in Air and Space Smithsonian Studies in History and Technology In these series, the Institution publishes small papers and full-scale monographs that report the research and collections of its various museums and bureaux or of professional colleagues in the world cf science and scholarship. The publications are distributed by mailing lists to libraries, universities, and similar institutions throughout the world. Papers or monographs submitted for series publication are received by the Smithsonian Institution Press, subject to its own review for format and style, only through departments of the various Smithsonian museums or bureaux, where the manuscripts are given substantive review. -

COMMANDE REF Désignation De L'article Taille QTE En Stock B00010 Three-Spot Angelfish Adult Apolemichthys Trimaculatus M 5 B005
QTE en COMMANDE REF Désignation de l'article Taille stock B00010 Three-spot Angelfish Adult Apolemichthys trimaculatus M 5 B00515 Bicolor Angelfish Centropyge bicolor M 35 B00530 Eibl's Angelfish Centropyge eibli M 13 B00540 White-tail Angelfish Centropyge flavicauda M 15 B00560 Midnight Angelfish Centropyge nox M 5 B00565 Keyhole Angelfish Centropyge tibicen M 5 B00570 Pearl-Scaled Angelfish Centropyge vroliki M 10 B010305 Yellowtail Vermiculated Angelfish Chaetodontoplus mesoleucus (Yellow) M 20 B02020 Emperor Angelfish Adult Pomacanthus imperator - M 10 B020205 Emperor Angelfish Juvenile Pomacanthus imperator (j) M 15 B02030 Blue-Girdled Angelfish Adult Pomacanthus navarchus - M 6 B02040 Koran Angelfish Adult Pomacanthus semicirculatus M 5 B020405 Koran Angelfish Juvenile Pomacanthus semicirculatus (j) M 6 B02050 Six-Banded Angelfish Adult Pomacanthus sexstriatus - M 5 B020505 Six-Banded Angelfish Juvenile Pomacanthus sexstriatus (j) M 5 B02060 Blue-Faced Angelfish Adult Pomacanthus xanthometopon - M 6 B020605 Blue-Faced Angelfish Juvenile Pomacanthus xanthometopon (j) M 5 B02510 Regal Angelfish Adult Pygoplites diacanthus - M 2 B04010 Longfin Bannerfish Heniochus acuminatus M 10 B04070 Humphead Bannerfish Heniochus varius M 2 B04510 Copperband Butterflyfish Chelmon rostratus M 150 B060110 Bantayan Butterflyfish Chaetodon adiergastos M 3 B060130 Threadfin Butterflyfish Chaetodon auriga M 2 B060140 Baroness Butterflyfish Chaetodon baronessa M 5 B060170 Citron Butterflyfish Chaetodon citrinellus M 5 B060190 Black-Finned Butterflyfish -

Reef Fishes of the Bird's Head Peninsula, West
Check List 5(3): 587–628, 2009. ISSN: 1809-127X LISTS OF SPECIES Reef fishes of the Bird’s Head Peninsula, West Papua, Indonesia Gerald R. Allen 1 Mark V. Erdmann 2 1 Department of Aquatic Zoology, Western Australian Museum. Locked Bag 49, Welshpool DC, Perth, Western Australia 6986. E-mail: [email protected] 2 Conservation International Indonesia Marine Program. Jl. Dr. Muwardi No. 17, Renon, Denpasar 80235 Indonesia. Abstract A checklist of shallow (to 60 m depth) reef fishes is provided for the Bird’s Head Peninsula region of West Papua, Indonesia. The area, which occupies the extreme western end of New Guinea, contains the world’s most diverse assemblage of coral reef fishes. The current checklist, which includes both historical records and recent survey results, includes 1,511 species in 451 genera and 111 families. Respective species totals for the three main coral reef areas – Raja Ampat Islands, Fakfak-Kaimana coast, and Cenderawasih Bay – are 1320, 995, and 877. In addition to its extraordinary species diversity, the region exhibits a remarkable level of endemism considering its relatively small area. A total of 26 species in 14 families are currently considered to be confined to the region. Introduction and finally a complex geologic past highlighted The region consisting of eastern Indonesia, East by shifting island arcs, oceanic plate collisions, Timor, Sabah, Philippines, Papua New Guinea, and widely fluctuating sea levels (Polhemus and the Solomon Islands is the global centre of 2007). reef fish diversity (Allen 2008). Approximately 2,460 species or 60 percent of the entire reef fish The Bird’s Head Peninsula and surrounding fauna of the Indo-West Pacific inhabits this waters has attracted the attention of naturalists and region, which is commonly referred to as the scientists ever since it was first visited by Coral Triangle (CT). -

SEASMART Program Final Report Annex
Creating a Sustainable, Equitable & Affordable Marine Aquarium Industry in Papua New Guinea | 1 Table of Contents Executive Summary ............................................................................................................ 7 Introduction ....................................................................................................................... 15 Contract Deliverables ........................................................................................................ 21 Overview of PNG in the Marine Aquarium Trade ............................................................. 23 History of the Global Marine Aquarium Trade & PNG ............................................ 23 Extent of the Global Marine Aquarium Trade .......................................................... 25 Brief History of Two Other Coastal Fisheries in PNG ............................................ 25 Destructive Potential of an Inequitable, Poorly Monitored & Managed Nature of the Trade Marine Aquarium Fishery in PNG ........................... 26 Benefit Potential of a Well Monitored & Branded Marine Aquarium Trade (and Other Artisanal Fisheries) in PNG ................................................................... 27 PNG Way to Best Business Practice & the Need for Effective Branding .............. 29 Economic & Environmental Benefits....................................................................... 30 Competitive Advantages of PNG in the Marine Aquarium Trade ................................... 32 Pristine Marine -

Training Manual Series No.15/2018
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by CMFRI Digital Repository DBTR-H D Indian Council of Agricultural Research Ministry of Science and Technology Central Marine Fisheries Research Institute Department of Biotechnology CMFRI Training Manual Series No.15/2018 Training Manual In the frame work of the project: DBT sponsored Three Months National Training in Molecular Biology and Biotechnology for Fisheries Professionals 2015-18 Training Manual In the frame work of the project: DBT sponsored Three Months National Training in Molecular Biology and Biotechnology for Fisheries Professionals 2015-18 Training Manual This is a limited edition of the CMFRI Training Manual provided to participants of the “DBT sponsored Three Months National Training in Molecular Biology and Biotechnology for Fisheries Professionals” organized by the Marine Biotechnology Division of Central Marine Fisheries Research Institute (CMFRI), from 2nd February 2015 - 31st March 2018. Principal Investigator Dr. P. Vijayagopal Compiled & Edited by Dr. P. Vijayagopal Dr. Reynold Peter Assisted by Aditya Prabhakar Swetha Dhamodharan P V ISBN 978-93-82263-24-1 CMFRI Training Manual Series No.15/2018 Published by Dr A Gopalakrishnan Director, Central Marine Fisheries Research Institute (ICAR-CMFRI) Central Marine Fisheries Research Institute PB.No:1603, Ernakulam North P.O, Kochi-682018, India. 2 Foreword Central Marine Fisheries Research Institute (CMFRI), Kochi along with CIFE, Mumbai and CIFA, Bhubaneswar within the Indian Council of Agricultural Research (ICAR) and Department of Biotechnology of Government of India organized a series of training programs entitled “DBT sponsored Three Months National Training in Molecular Biology and Biotechnology for Fisheries Professionals”. -

Zoo Keeper Information
ZOO KEEPER INFORMATION Auckland Zoo and its role in Conservation and Captive Breeding Programmes Revised by Kirsty Chalmers Registrar 2006 CONTENTS Introduction 3 Auckland Zoo vision, mission and strategic intent 4 The role of modern zoos 5 Issues with captive breeding programmes 6 Overcoming captive breeding problems 7 Assessing degrees of risk 8 IUCN threatened species categories 10 Trade in endangered species 12 CITES 12 The World Zoo and Aquarium Conservation Strategy 13 International Species Information System (ISIS) 15 Animal Records Keeping System (ARKS) 15 Auckland Zoo’s records 17 Identification of animals 17 What should go on daily reports? 18 Zoological Information Management System (ZIMS) 19 Studbooks and SPARKS 20 Species co-ordinators and taxon advisory groups 20 ARAZPA 21 Australasian Species Management Program (ASMP) 21 Animal transfers 22 Some useful acronyms 24 Some useful references 25 Appendices 26 Zoo Keeper Information 2006 2 INTRODUCTION The intention of this manual is to give a basic overview of the general operating environment of zoos, and some of Auckland Zoo’s internal procedures and external relationships, in particular those that have an impact on species management and husbandry. The manual is designed to be of benefit to all keepers, to offer a better understanding of the importance of captive animal husbandry and species management on a national and international level. Zoo Keeper Information 2006 3 AUCKLAND ZOO VISION Auckland Zoo will be globally acknowledged as an outstanding, progressive zoological park. AUCKLAND ZOO MISSION To focus the Zoo’s resources to benefit conservation and provide exciting visitor experiences which inspire and empower people to take positive action for wildlife and the environment. -

Fish 10000 Genomes Project
bioRxiv preprint doi: https://doi.org/10.1101/787028; this version posted September 30, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Initial data release and announcement of the Fish10K: Fish 10,000 Genomes Project Guanngyi Fan1,5,*, Yue Song1,*, Xiaoyun Huang1,*, Liandong Yang2,*, Suyu Zhang1, Mengqi Zhang1, Xianwei Yang1, Yue Chang1, He Zhang1,5, Yongxin Li3, Shanshan Liu1, Lili Yu1, Inge Seim8,9, Chenguang Feng3, Wen Wang3, Kun Wang3, Jing Wang4,6,7, Xun Xu5, Huanming Yang1,5, Nansheng Chen4,6,7,†, Xin Liu1,5,† & Shunping He2,†. 1BGI-Qingdao, BGI-Shenzhen, Qingdao 266555, China 2Key Laboratory of Aquatic Biodiversity and Conservation, Institute of Hydrobiology, Chinese Academy of Sciences, Wuhan, China 3Center for Ecological and Environmental Sciences, Northwestern Polytechnical University, Xi’an, China. 4CAS Key Laboratory of Marine Ecology and Environmental Sciences, Institute of Oceanology, Chinese Academy of Sciences, Qingdao, Shandong 266071, China 5BGI-Shenzhen, Shenzhen 518083, China 6Marine Ecology and Environmental Science Laboratory, Pilot National Laboratory for Marine Science and Technology (Qingdao), Qingdao, Shandong 266237, PR China 7Center for Ocean Mega-Science, Chinese Academy of Sciences, Qingdao, Shandong 266400, PR China 8Integrative Biology Laboratory, College of Life Sciences, Nanjing Normal University, Nanjing 210023, China; 9Comparative and Endocrine Biology Laboratory, Translational Research Institute-Institute of Health and Biomedical Innovation, School of Biomedical Sciences, Queensland University of Technology, Brisbane 4102, Queensland, 1 bioRxiv preprint doi: https://doi.org/10.1101/787028; this version posted September 30, 2019. -

Ecological and Life History Factors Influence Habitat and Tool Use in Wild Bottlenose Dolphins (Tursiops Sp.)
ECOLOGICAL AND LIFE HISTORY FACTORS INFLUENCE HABITAT AND TOOL USE IN WILD BOTTLENOSE DOLPHINS (TURSIOPS SP.) A Dissertation submitted to the Faculty of the Graduate School of Arts and Sciences of Georgetown University in partial fulfillment of the requirements for the degree of Doctor of Philosophy in Biology By Eric Michael Patterson, B.S. Washington, DC December 7, 2012 Copyright 2011 by Eric Michael Patterson All Rights Reserved ii ECOLOGICAL AND LIFE HISTORY FACTORS INFLUENCE HABITAT AND TOOL USE IN WILD BOTTLENOSE DOLPHINS (TURSIOPS SP.) Eric Michael Patterson, B.S. Thesis Advisor: Janet Mann, Ph.D. ABSTRACT While it has long been known that individual animals behave quite distinctively from other conspecifics, only recently has this intraspecific behavioral variation itself been the subject of investigation rather than a nuisance in statistical analyses. As research amounts, it is becoming ever more apparent that the ecology of individuals can have real and measurable biological consequences. Previous studies have documented pronounced inter-individual variation in foraging behaviors in Shark Bay wild bottlenose dolphins, which often coincide with individual specialization. In this dissertation I examine how individuals’ phenotypic characteristics relate to their foraging behavior, habitat use, and ranging. In Chapter 1 I explore habitat and ranging for adult male and female bottlenose dolphins. Dolphins show marked intraspecific variation in both habitat use and ranging, which partially relates to foraging tactic, sex, and season. However, some variation cannot be explained by any obvious phenotypic or ecological factors, highlighting the importance of individual level analyses, especially for conservation and management. In Chapter 2 I examine the underlying basis of one foraging tactic involving tool use: sponge foraging. -

Evolutionary History of the Butterflyfishes (F: Chaetodontidae
doi:10.1111/j.1420-9101.2009.01904.x Evolutionary history of the butterflyfishes (f: Chaetodontidae) and the rise of coral feeding fishes D. R. BELLWOOD* ,S.KLANTEN*à,P.F.COWMAN* ,M.S.PRATCHETT ,N.KONOW*§ &L.VAN HERWERDEN*à *School of Marine and Tropical Biology, James Cook University, Townsville, Qld, Australia Australian Research Council Centre of Excellence for Coral Reef Studies, James Cook University, Townsville, Qld, Australia àMolecular Evolution and Ecology Laboratory, James Cook University, Townsville, Qld, Australia §Ecology and Evolutionary Biology, Brown University, Providence, RI, USA Keywords: Abstract biogeography; Of the 5000 fish species on coral reefs, corals dominate the diet of just 41 chronogram; species. Most (61%) belong to a single family, the butterflyfishes (Chae- coral reef; todontidae). We examine the evolutionary origins of chaetodontid corallivory innovation; using a new molecular phylogeny incorporating all 11 genera. A 1759-bp molecular phylogeny; sequence of nuclear (S7I1 and ETS2) and mitochondrial (cytochrome b) data trophic novelty. yielded a fully resolved tree with strong support for all major nodes. A chronogram, constructed using Bayesian inference with multiple parametric priors, and recent ecological data reveal that corallivory has arisen at least five times over a period of 12 Ma, from 15.7 to 3 Ma. A move onto coral reefs in the Miocene foreshadowed rapid cladogenesis within Chaetodon and the origins of corallivory, coinciding with a global reorganization of coral reefs and the expansion of fast-growing corals. This historical association underpins the sensitivity of specific butterflyfish clades to global coral decline. butterflyfishes (f. Chaetodontidae); of the remainder Introduction most (eight) are in the Labridae. -

Hidden Havens: Exploring Marine Life in Singapore's Marinas
HIDDEN HAVENS HIDDEN EXPLORING Marine Life IN SINGAPORE’S EXPLORING Marine Life IN SINGAPORE’S MARINAS MARINAS In collaboration with: Chou Loke Ming, Lionel Ng, Toh Kok Ben, Cheo Pei Rong, Ng Juat Ying and Karenne Tun 9 789811 452611 HIDDEN HAVENS HIDDEN EXPLORING Marine Life IN SINGAPORE’S MARINAS EXPLORING Marine Chou Loke Ming, Lionel Ng, Life IN SINGAPORE’S Toh Kok Ben, Cheo Pei Rong, EXPLORING Marine Life IN SINGAPORE’S MARINAS MARINAS Ng Juat Ying and Karenne Tun In collaboration with: Chou Loke Ming, Lionel Ng, Toh Kok Ben, Cheo Pei Rong, Ng Juat Ying and Karenne Tun 9 789811 452581 Cuttlefishes, cardinalfishes, fan worms, seafans and sea anemones are just some of the fascinating sea creatures that you may find in Singapore’s marinas. Beneath the surface of our urban waters, a myriad of forms and colours can In collaboration with: be found in these hidden havens of biodiversity. 2 HIDDEN HAVENS EXPLORING Marine Life IN SINGAPORE’S MARINAS 3 FOREWORD At first glance, a casual observer may think that Singapore’s marine environment is devoid of life, just because we are one of the world’s busiest ports. Many people will therefore be surprised to learn that our coastal waters are home to a rich and diverse selection of marine life, even in highly-modified environments like the marinas. Hidden Havens: Exploring Marine Life in Singapore’s Marinas is an aptly named book that will hopefully bring readers on a journey of discovery as we explore the myriad of living things that thrive in our marinas. -
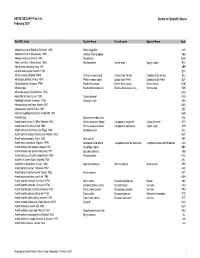
ASFIS ISSCAAP Fish List February 2007 Sorted on Scientific Name
ASFIS ISSCAAP Fish List Sorted on Scientific Name February 2007 Scientific name English Name French name Spanish Name Code Abalistes stellaris (Bloch & Schneider 1801) Starry triggerfish AJS Abbottina rivularis (Basilewsky 1855) Chinese false gudgeon ABB Ablabys binotatus (Peters 1855) Redskinfish ABW Ablennes hians (Valenciennes 1846) Flat needlefish Orphie plate Agujón sable BAF Aborichthys elongatus Hora 1921 ABE Abralia andamanika Goodrich 1898 BLK Abralia veranyi (Rüppell 1844) Verany's enope squid Encornet de Verany Enoploluria de Verany BLJ Abraliopsis pfefferi (Verany 1837) Pfeffer's enope squid Encornet de Pfeffer Enoploluria de Pfeffer BJF Abramis brama (Linnaeus 1758) Freshwater bream Brème d'eau douce Brema común FBM Abramis spp Freshwater breams nei Brèmes d'eau douce nca Bremas nep FBR Abramites eques (Steindachner 1878) ABQ Abudefduf luridus (Cuvier 1830) Canary damsel AUU Abudefduf saxatilis (Linnaeus 1758) Sergeant-major ABU Abyssobrotula galatheae Nielsen 1977 OAG Abyssocottus elochini Taliev 1955 AEZ Abythites lepidogenys (Smith & Radcliffe 1913) AHD Acanella spp Branched bamboo coral KQL Acanthacaris caeca (A. Milne Edwards 1881) Atlantic deep-sea lobster Langoustine arganelle Cigala de fondo NTK Acanthacaris tenuimana Bate 1888 Prickly deep-sea lobster Langoustine spinuleuse Cigala raspa NHI Acanthalburnus microlepis (De Filippi 1861) Blackbrow bleak AHL Acanthaphritis barbata (Okamura & Kishida 1963) NHT Acantharchus pomotis (Baird 1855) Mud sunfish AKP Acanthaxius caespitosa (Squires 1979) Deepwater mud lobster Langouste -

Les Genres Et Sous-Genres De Chaetodontidés Étudiés Par Une Méthode D'analyse Numérique
Bull. Mus. natn. Hist, nat., Paris, 4e sér., 6, 1984, section A, n° 2 : 453-485. Les genres et sous-genres de Chaetodontidés étudiés par une méthode d'analyse numérique par André MAUGÉ et Roland BAUCHOT Résumé. — L'analyse en composantes principales des cent quinze espèces de Chaetodontidae à l'aide de trente variables (neuf valeurs méristiques, treize proportions de diverses parties du corps et huit caractères morphologiques) a permis de préciser, à partir de divers dendrogrammes, les affinités de ces espèces entre elles et de proposer une nouvelle répartition des Chaetodontidae en vingt genres, dont quatre nouveaux, et vingt et un sous-genres, dont six nouveaux. Abstract. — Principal component analysis of the 115 species of Chaetodontids using 30 varia- bles (9 meristic data, 13 proportions of different body parts and 8 morphological characters) allowed us to define, from the study of different dendrograms, the affinities of the species and to propose a new classification of the Chaetodontids in 20 genera (4 of which are new) and 21 sub-genera (6 of which are new). A. MAUGÉ, Laboratoire d'Ichtyologie générale et appliquée, Muséum national d'Histoire naturelle, 43, rue Cuvier, 75231 Paris cedex 05. R. BAUCHOT, Laboratoire d'Anatomie comparée, Université Paris VII, 2, place Jussieu, 75221 Paris cedex 05. La famille des chétodons (Chaetodontidae) a toujours formé un puzzle, dont tous les auteurs qui ont eu à en connaître depuis BLEEKER se sont efforcés de classer les éléments et d'harmoniser les sous-classements. Certains ont tenté cet effort pour une aire géogra- phique restreinte, fonction des limites des faunes envisagées.