Development and Validation of an Ultra-Fast and Sensitive Microflow
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Stability Study of Cannabidiol in the Form of Solid Powder and Sunflower Oil Solution
pharmaceutics Article Stability Study of Cannabidiol in the Form of Solid Powder and Sunflower Oil Solution Ema Kosovi´c 1,2 , David Sýkora 2 and Martin Kuchaˇr 3,* 1 Institute of Chemical Process Fundamentals of CAS v.v.i., Rozvojová 135, 16502 Prague, Czech Republic; [email protected] 2 Department of Analytical Chemistry, University of Chemistry and Technology Prague, Technická 5, 16628 Prague, Czech Republic; [email protected] 3 Forensic Laboratory of Biologically Active Substances, Department of Chemistry of Natural Compounds, University of Chemistry and Technology Prague, Technická 5, 16628 Prague, Czech Republic * Correspondence: [email protected] Abstract: Stability studies represent an essential component of pharmaceutical development, en- abling critical evaluation of the therapeutic potential of an active pharmaceutical ingredient (API) or a final pharmaceutical product under the influence of various environmental factors. The aim of the present study was to investigate the chemical stability of cannabidiol (CBD) in the form of a solid powder (hereinafter referred to as CBD powder) and also dissolved in sunflower oil. We performed stress studies in accordance with the International Conference on Harmonization (ICH) guidelines, where 5 mg of marketed CBD in the form of a solid powder and in form of oil solution were exposed for 7 and 14, 30, 60, 90, 180, 270, and 365 days to precisely defined temperature and humidity conditions, 25 ◦C ± 2 ◦C/60% RH ± 5% and 40 ◦C ± 2 ◦C/75% RH ± 5% in both open and closed vials in the dark. CBD powder was significantly more stable than CBD in oil solution. Such finding is important because CBD is often administered dissolved in oil matrix in practice due to Citation: Kosovi´c,E.; Sýkora, D.; very good bioavailability. -

Endocannabinoid Stimulated Release of Nitric Oxide and Its Mitochondrial
A tica nal eu yt c ic a a m A r a c t Stefano et al., Pharm Anal Acta 2015, 6:6 h a P Pharmaceutica Analytica Acta DOI: 10.4172/2153-2435.1000378 ISSN: 2153-2435 Review Article Open Access Endocannabinoid Stimulated Release of Nitric Oxide and its Mitochondrial Influence Triggering Vascular Pathology George B Stefano*, Erin Quinn and Richard M Kream MitoGenetics LLC, 3 Bioscience Park Drive, Suite 307, Farmingdale, NY 11735, USA Abstract Endocannabinoids, and their respective receptors, are involved in a host of cellular regulatory activities. In part, some of these mediated effects occur by way of stimulating constitutive nitric oxide release. This occurs in endothelia, certain white blood cells, microglia, and in similar invertebrate tissues, demonstrating that this is a conserved chemical messenger system. This endocannabinoid chemical messenger system, coupled to constitutive nitric oxide release, also appears to exert regulatory effects on mitochondrial energy associated processes, further substantiating its primordial history. In this regard, it appears to offer some beneficial actions in the occurrence of reperfusion injury and stroke. The mechanism envisioned is one initiated via a hypoxic event, which does not restore normalcy, then progresses to a pro-inflammatory state, and the resultant chronic condition manifests itself in a specific disorder. This fits nicely into a vascular-associated origin for Alzheimer’s Disease, whereby the pro- inflammatory state encompasses vessels that have endothelial gaps, providing for a compromised blood brain barrier, beta amyloid deposition, and enhanced white blood cell trafficking. In time, due to the physical progression of the events, Alzheimer’s Disease occurs. -

FDA Regulation of Cannabidiol (CBD) Products
June 12, 2019 FDA Regulation of Cannabidiol (CBD) Products Cannabidiol (CBD) is promoted as treatment for a range of and hemp-derived compounds, including CBD. However, conditions, including epileptic seizures, post-traumatic the farm bill explicitly preserved FDA’s authority under the stress disorder, anxiety, and inflammation—despite limited Federal Food, Drug and Cosmetic Act (FFDCA, 21 U.S.C. scientific evidence to substantiate many of these claims. In §§301 et seq.) and Section 351 of the Public Health Service the United States, CBD is marketed in food and beverages, Act (PHSA, 42 U.S.C. §262), including for hemp-derived dietary supplements, and cosmetics—products that are products. According to FDA, “because the 2018 Farm Bill regulated by the Food and Drug Administration (FDA). did not change FDA’s authorities, cannabis and cannabis- CBD is also the active ingredient in an FDA-approved pharmaceutical drug, Epidiolex®. CBD is a plant-derived derived products are subject to the same authorities and substance from Cannabis sativa, the species of plant that requirements as FDA-regulated products containing any includes both hemp and marijuana, but from different plant other substance, regardless of whether the products fall varieties or cultivars. CBD is the primary nonpsychoactive within the definition of ‘hemp’ under the 2018 Farm Bill.” compound in cannabis, whereas tetrahydrocannabinol (THC) is cannabis’s primary psychoactive compound. FDA Regulation of CBD Products FDA, under the FFDCA, regulates many of the products Regulation of CBD Products marketed as containing cannabis and cannabis-derived Hemp and marijuana have separate definitions in U.S. law compounds, including CBD. -

NIDA Drug Supply Program Catalog, 25Th Edition
RESEARCH RESOURCES DRUG SUPPLY PROGRAM CATALOG 25TH EDITION MAY 2016 CHEMISTRY AND PHARMACEUTICS BRANCH DIVISION OF THERAPEUTICS AND MEDICAL CONSEQUENCES NATIONAL INSTITUTE ON DRUG ABUSE NATIONAL INSTITUTES OF HEALTH DEPARTMENT OF HEALTH AND HUMAN SERVICES 6001 EXECUTIVE BOULEVARD ROCKVILLE, MARYLAND 20852 160524 On the cover: CPK rendering of nalfurafine. TABLE OF CONTENTS A. Introduction ................................................................................................1 B. NIDA Drug Supply Program (DSP) Ordering Guidelines ..........................3 C. Drug Request Checklist .............................................................................8 D. Sample DEA Order Form 222 ....................................................................9 E. Supply & Analysis of Standard Solutions of Δ9-THC ..............................10 F. Alternate Sources for Peptides ...............................................................11 G. Instructions for Analytical Services .........................................................12 H. X-Ray Diffraction Analysis of Compounds .............................................13 I. Nicotine Research Cigarettes Drug Supply Program .............................16 J. Ordering Guidelines for Nicotine Research Cigarettes (NRCs)..............18 K. Ordering Guidelines for Marijuana and Marijuana Cigarettes ................21 L. Important Addresses, Telephone & Fax Numbers ..................................24 M. Available Drugs, Compounds, and Dosage Forms ..............................25 -

CBD) Information About CBD in Cannabis and Hemp Products Under the New Cannabis Act
ALL ABOUT CANNABIDIOL (CBD) Information about CBD in cannabis and hemp products under the new Cannabis Act WHAT IS CBD? The cannabis plant contains hundreds of different chemical constituents, such as cannabinoids, terpenes and flavonoids. Currently, over 100 chemical substances collectively known as cannabinoids have been identified. Cannabinoids that are derived from cannabis plants are sometimes referred to as phytocannabinoids. Cannabidiol (CBD) is one of these cannabinoids. Unlike tetrahydrocannabinol (THC) - the most well-known cannabinoid in cannabis, responsible for the high or intoxication of consuming cannabis - CBD does not produce a high or intoxicating effect. However, there is some evidence that CBD may influence some of the effects that THC has on the brain. CBD is also being studied for its possible therapeutic uses; it has received market approval in the United States for two severe forms of epilepsy. CBD can be found in varieties of the cannabis plant, including hemp plants. All phytocannabinoids, whether derived from a cannabis plant or produced by synthesis, including both THC and CBD, are regulated under the new Cannabis Act and its regulations, which came into force on October 17, 2018. HOW IS CBD REGULATED IN CANADA? Under the Cannabis Act, many activities with phytocannabinoids, CBD included, remain prohibited, except for the specific cases authorized by the Act and its regulations, which include strict controls on possession, production, sale, and distribution. While Health Canada oversees the production of cannabis products, the provinces and territories oversee the distribution and retail aspects of the cannabis supply chain. Health Canada remains responsible for overseeing the distribution and sale of cannabis and any CBD-containing cannabis products for medical purposes. -

Cannabidiol and ( ) 9-Tetrahydrocannabinol Are Neuroprotective Antioxidants
Proc. Natl. Acad. Sci. USA Vol. 95, pp. 8268–8273, July 1998 Medical Sciences Cannabidiol and (2)D9-tetrahydrocannabinol are neuroprotective antioxidants A. J. HAMPSON*†,M.GRIMALDI‡,J.AXELROD*, AND D. WINK§ *Laboratory of Cellular and Molecular Regulation, National Institutes of Mental Health, Bethesda, MD 20892; ‡Laboratory of Adaptive Systems, National Institute of Neurological Disorders and Stroke, Bethesda, MD 20892; and §Radiology and Biology Branch, National Cancer Institute, Bethesda, MD 20892 Contributed by Julius Axelrod, April 27, 1998 ABSTRACT The neuroprotective actions of cannabidiol reduce the toxic reactive oxygen species (ROS) formed during and other cannabinoids were examined in rat cortical neuron ischemic metabolism. Cannabinoids like (2)D9-tetrahydrocan- cultures exposed to toxic levels of the excitatory neurotrans- nabinol (THC) and its psychoactive analogues also have been mitter glutamate. Glutamate toxicity was reduced by both reported to be neuroprotective against glutamate toxicity in cannabidiol, a nonpsychoactive constituent of marijuana, and vitro (7). Cannabinoids have been suggested to prevent gluta- the psychotropic cannabinoid (2)D9-tetrahydrocannabinol mate neurotoxicity by activating cannabinoid receptors (7, 8), (THC). Cannabinoids protected equally well against neuro- which can reduce calcium influx through voltage sensitive toxicity mediated by N-methyl-D-aspartate receptors, 2-ami- calcium channels (8, 9). A synthetic cannabinoid (HU-211) no-3-(4-butyl-3-hydroxyisoxazol-5-yl)propionic acid recep- also has been demonstrated to be neuroprotective even though tors, or kainate receptors. N-methyl-D-aspartate receptor- it does not activate cannabinoid receptors. This compound is induced toxicity has been shown to be calcium dependent; this an atypical cannabinoid, however, in that it, unlike other study demonstrates that 2-amino-3-(4-butyl-3-hydroxyisox- cannabinoids, directly antagonizes NMDAr (10) and possesses azol-5-yl)propionic acidykainate receptor-type neurotoxicity some antioxidant properties (11). -

Selected Articles on the Analysis of Drugs of Abuse in Seized Materials (January-June 2017) Differentiation of Ring-Substituted
Selected Articles on the Analysis of Drugs of Abuse in Seized Materials (January-June 2017) Differentiation of ring-substituted regioisomers of amphetamine and methamphetamine by supercritical fluid chromatography, H. Segawa, Y. T. Iwata, T. Yamamuro, K. Kuwayama, K. Tsujikawa, T. Kanamori and H. Inoue, Drug Testing and Analysis, 2017, 9, 389-398. http://dx.doi.org/10.1002/dta.2040 Rapid detection of fentanyl, fentanyl analogues, and opioids for on-site or laboratory based drug seizure screening using thermal desorption DART-MS and ion mobility spectrometry, E. Sisco, J. Verkouteren, J. Staymates and J. Lawrence, Forensic Chemistry, 2017, 4, 108-115. http://www.sciencedirect.com/science/article/pii/S2468170917300152 Detection of phenazepam in illicitly manufactured Erimin 5 tablets, W. J. L. Lim, A. T. W. Yap, M. Mangudi, H. B. Koh, A. S. Y. Tang and K. B. Chan, Drug Testing and Analysis, 2017, 9, 293-305. http://dx.doi.org/10.1002/dta.1981 Development of a new field-test procedure for cocaine, K. Tsujikawa, Y. T. Iwata, H. Segawa, T. Yamamuro, K. Kuwayama, T. Kanamori and H. Inoue, Forensic Science International, 2017, 270, 267-274. https://www.sciencedirect.com/science/article/pii/S0379073816304662 Quantitative Determination of Cannabinoids in Cannabis and Cannabis Products Using Ultra-High- Performance Supercritical Fluid Chromatography and Diode Array/Mass Spectrometric Detection, M. Wang, Y.-H. Wang, B. Avula, M. M. Radwan, A. S. Wanas, Z. Mehmedic, J. van Antwerp, M. A. ElSohly and I. A. Khan, J Forensic Sci, 2017, 62, 602-611. http://onlinelibrary.wiley.com.ezproxyhost.library.tmc.edu/doi/10.1111/1556-4029.13341/abstract Selected Articles on the Analysis of Drugs of Abuse in Biological Specimens (January-June 2017) Simultaneous quantification of major cannabinoids and metabolites in human urine and plasma by HPLC-MS/MS and enzyme-alkaline hydrolysis, O. -
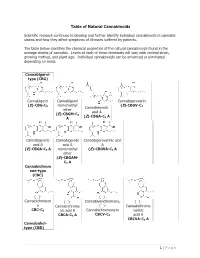
Table of Natural Cannabinoids
Table of Natural Cannabinoids Scientific research continues to develop and further identify individual cannabinoids in cannabis strains and how they affect symptoms of illnesses suffered by patients. The table below identifies the chemical properties of the natural cannabinoids found in the average strains of cannabis. Levels of each of these chemicals will vary with varietal strain, growing method, and plant age. Individual cannabinoids can be enhanced or eliminated depending on need. Cannabigerol- type (CBG) Cannabigerol Cannabigerol Cannabigerovarin (E)-CBG-C monomethyl (E)-CBGV-C 5 Cannabinerolic 3 ether acid A (E)-CBGM-C 5 (Z)-CBGA-C A A 5 Cannabigerolic Cannabigerolic Cannabigerovarinic acid acid A acid A A (E)-CBGA-C5 A monomethyl (E)-CBGVA-C3 A ether (E)-CBGAM- C5 A Cannabichrom ene-type (CBC) (±)- (±)- Cannabichromen (±)- Cannabivarichromene, (±)- e Cannabichrome (±)- Cannabichrome CBC-C5 nic acid A Cannabichromevarin varinic CBCA-C5 A CBCV-C3 acid A CBCVA-C3 A Cannabidiol- type (CBD) 1 | Page (−)-Cannabidiol Cannabidiol Cannabidiol-C4 (−)- Cannabidiorc CBD-C5 momomethyl CBD-C4 Cannabidivarin ol ether CBDV-C3 CBD-C1 CBDM-C5 Cannabidiolic Cannabidivarini acid c acid CBDA-C5 CBDVA-C3 Cannabinodiol- type (CBND) Cannabinodiol Cannabinodivar CBND-C5 in CBND-C3 Tetrahydrocan nabinol-type (THC) 9 9 9 Δ - Δ - Δ - Δ9- Tetrahydrocanna Tetrahydrocan Tetrahydrocannabivarin 9 Tetrahydrocan binol nabinol-C4 Δ -THCV-C3 9 9 nabiorcol Δ -THC-C5 Δ -THC-C4 9 Δ -THCO-C1 9 9 Δ -Tetrahydro- Δ9-Tetrahydro- Δ -Tetrahydro- Δ9-Tetrahydro- cannabinolic -

Read the Latest Analysis of the Bill Here
SB 519 Page 1 Date of Hearing: July 13, 2021 ASSEMBLY COMMITTEE ON HEALTH Jim Wood, Chair SB 519 (Wiener) – As Amended July 1, 2021 AS PROPOSED TO BE AMENDED SENATE VOTE: 21-16 SUBJECT: Controlled substances: decriminalization of certain hallucinogenic substances. SUMMARY: Makes it lawful for a person 21 years of age or older to possess, obtain, and ingest specified controlled substances classified as hallucinogens. Makes it lawful for the facilitated or supportive use of specified controlled substances classified as hallucinogens. Makes it unlawful for persons under the age of 21 to do such acts related to those hallucinogens, as specified. Requires the California Department of Public Health (DPH) to convene a working group to study specified hallucinogens and make recommendations to the Legislature regarding the statewide regulation of those hallucinogens and other controlled substances. Specifically, this bill: 1) Defines mescaline as derived from plants presently classified botanically in the Echinopsis or Trichocereus genus of cacti, including, without limitation, the Bolivian Torch Cactus, San Pedro Cactus, or Peruvian Torch Cactus, but not including mescaline derived from any plant defined as peyote. 2) Provides that, if there is a change in federal law permitting the prescription, furnishing, or dispensing of psilocybin, psilocyn, dimethyltryptamine (DMT), Ibogaine, mescaline, lysergic acid diethylamide (LSD), or 3,4-methylenedioxymethamphetamine (MDMA) products, a physician, pharmacist, or other authorized healing arts licensee acting within their scope of practice who prescribes, furnishes, or dispenses that product in accordance with federal law, shall be deemed to be in compliance with state law. 3) Removes mescaline from existing law that prohibits the possession of specified controlled substances. -

Minireview the Pharmacological Case for Cannabigerol
1521-0103/376/2/204–212$35.00 https://doi.org/10.1124/jpet.120.000340 THE JOURNAL OF PHARMACOLOGY AND EXPERIMENTAL THERAPEUTICS J Pharmacol Exp Ther 376:204–212, February 2021 Copyright ª 2021 by The Author(s) This is an open access article distributed under the CC BY-NC Attribution 4.0 International license. Minireview The Pharmacological Case for Cannabigerol Rahul Nachnani, Wesley M. Raup-Konsavage, and Kent E. Vrana Department of Pharmacology, Penn State College of Medicine, Hershey, Pennsylvania Received September 15, 2020; accepted November 4, 2020 ABSTRACT Downloaded from Medical cannabis and individual cannabinoids, such as D9- antibacterial activity. There is growing interest in the commercial tetrahydrocannabinol (D9-THC) and cannabidiol (CBD), are re- use of this unregulated phytocannabinoid. This review focuses ceiving growing attention in both the media and the scientific on the unique pharmacology of CBG, our current knowledge literature. The Cannabis plant, however, produces over 100 of its possible therapeutic utility, and its potential toxicolog- different cannabinoids, and cannabigerol (CBG) serves as the ical hazards. precursor molecule for the most abundant phytocannabinoids. jpet.aspetjournals.org CBG exhibits affinity and activity characteristics between SIGNIFICANCE STATEMENT D9-THC and CBD at the cannabinoid receptors but appears Cannabigerol is currently being marketed as a dietary supple- to be unique in its interactions with a-2 adrenoceptors and ment and, as with cannabidiol (CBD) before, many claims are 5-hydroxytryptamine (5-HT1A). Studies indicate that CBG may being made about its benefits. Unlike CBD, however, little have therapeutic potential in treating neurologic disorders research has been performed on this unregulated molecule, (e.g., Huntington disease, Parkinson disease, and multiple and much of what is known warrants further investigation to sclerosis) and inflammatory bowel disease, as well as having identify potential areas of therapeutic uses and hazards. -

Recreational Drugs: Amyl Nitrate, Amphetamines, Cannabis, Cocaine/Crack, Ghb, Hallucinogens, Ketamine, Heroin, Naloxone
RECREATIONAL DRUGS: AMYL NITRATE, AMPHETAMINES, CANNABIS, COCAINE/CRACK, GHB, HALLUCINOGENS, KETAMINE, HEROIN, NALOXONE INSTIs NNRTIs PIs • BICTEGRAVIR (Biktarvy) • ELVITEGRAVIR/ • DORAVIRINE (Pifeltro, • EFAVIRENZ (Sustiva, Boosted with ritonavir • DOLUTEGRAVIR COBICISTAT (Stribild, Delstrigo) Atripla) (Norvir) or cobicistat (Tivicay, Triumeq, Genvoya) • RILPIVIRINE (Edurant, • ETRAVIRINE (Intelence) • ATAZANAVIR (Reyataz, Juluca) Complera, Odefsey, • NEVIRAPINE Evotaz) • RALTEGRAVIR Juluca) (Viramune) • DARUNAVIR (Prezista, (Isentress) Prezcobix, Symtuza) • LOPINAVIR (Kaletra) AMYL NITRATE • Poppers, ames AMPHETAMINES • MDMA/ecstasy, Potential for ↑ Potential for ↑ recreational crystal, molly recreational drug drug CANNABIS (THC), CANNABIDIOL (CBD) *Note that oral cannabis oils or dried cannabis may include THC/CBD in various ratios • Marijuana, weed Potential for ↑ THC & CBD Potential for ↑ THC and Potential for ↑ THC ↓ CBD Potential for ↑/↓ CBD COCAINE • crack, base Potential for ↑ Potential QT prolongation Potential for ↑ levels of Potential for ↑ recreational recreational drug (rilpivirine) hepatotoxic metabolite drug GAMMA-HYDROXYBUTYRATE • GHB, date rape Potential for ↑ Potential for ↑ recreational drug, Geeb, liquid X recreational drug drug INSTIs NNRTIs PIs • BICTEGRAVIR (Biktarvy) • ELVITEGRAVIR/ • DORAVIRINE (Pifeltro, • EFAVIRENZ (Sustiva, Boosted with ritonavir • DOLUTEGRAVIR COBICISTAT (Stribild, Delstrigo) Atripla) (Norvir) or cobicistat (Tivicay, Triumeq, Genvoya) • RILPIVIRINE (Edurant, • ETRAVIRINE (Intelence) • ATAZANAVIR -

The Effects of Cannabidiol-Based Sublingual Tablets on Diabetic Neuropathic Pain Debra Kimless, M.D., Matthew K
abetes & OPEN ACCESS Freely available online Di M f e t o a l b a o n l r i s u m o J Journal of Diabetes & Metabolism ISSN: 2155-6156 Research Article The Effects of Cannabidiol-Based Sublingual Tablets on Diabetic Neuropathic Pain Debra Kimless, M.D., Matthew K. Caloura*, Ara Kirakosyan, Ph.D., and Stephen Goldner, Esq Pure Green Pharmaceuticals, West Bloomfield, 48323, USA ABSTRACT Background: This is a prospective, open-label, drug and dose-controlled study. The primary objective of this study is to evaluate the safety and efficacy of cannabidiol (CBD) sublingual tablets for the treatment of patients with chronic diabetic neuropathic pain. The secondary objects are to evaluate the impact of CBD tablets on sleep quality and anxiety. We describe the effectiveness data at the completion of a 21-day study. Methods: Subjects who met all inclusion/exclusion criteria and met the prespecified minimum Numerical Pain Rating Scale (NPRS) score were eligible for participation. Thirty-one subjects with chronic diabetic neuropathic pain were recruited. Patients were instructed to take CBD sublingual tablets three times a day and were taught to use a smart phone data collection application to record daily NPRS scores. Results: At the conclusion of the study, patients reported significant reduction in pain from baseline. The secondary endpoints analysing the effects of the CBD tablets on sleep quality and anxiety score revealed statistically significant improvement in both. No adverse drug reactions were reported. Conclusions: Our findings demonstrate that the sublingual application of CBD tablets can achieve significant reduction in pain, as well as significant improvement in sleep quality and a reduction in anxiety and without any adverse drug reactions in patients with diabetic peripheral neuropathy.