Development and Validation of an LC-MS/MS Method After Chiral Derivatization for the Simultaneous Stereoselective Determination of Methylenedioxy-Methamphetamine
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

(Mdma) in Non Conventional Matrices and Its Applications in Clinical Toxicology
Doctoral Thesis Doctorat Farmacología Departament de Farmacología, de Terapéutica i de Toxicologia DISTRIBUTION OF 3,4-METHYLENEDIOXYMETHAMPHETAMINE (MDMA) IN NON CONVENTIONAL MATRICES AND ITS APPLICATIONS IN CLINICAL TOXICOLOGY SIMONA PICHINI p Doctoral Thesis Doctorat Farmacología Departament de Farmacología, de Terapéutica i de Toxicologia DISTRIBUTION OF 3,4-METHYLENEDIOXYMETHAMPHETAMINE (MDMA) IN NON CONVENTIONAL MATRICES AND ITS APPLICATIONS IN CLINICAL TOXICOLOGY Doctoral thesis submitted by Simona Pichini as a partial fulfillment of the requirements for the degree of Doctor by the Universitat Autònoma de Barcelona. The studies included in this thesis have been realized under the direction of Dr. Rafael de la Torre Fornell and Dr. Magí Farré Albaladejo at the Pharmacology Unit of the Institut Municipal d'Investigació Mèdica (IMIM), Barcelona, Spain; and at the Drug Research and Control Department of the Istituto Superiore di Sanità, Roma, Italy. Doctorate Program of the Universitat Autònoma de Barcelona. Signature of Thesis Director Signature of Thesis Director (Dr. Magí Farré Albaladejo) (Dr. Rafael de la Torre Fornell) Signature of Doctorand (Simona Pichini) Yo soy yo y aquéllos a quienes amo Jorge Bucay To my friends, treasure of my life Acknowledgements Acknowledgements A number of people contributed to high extent to achieve the objectives of this Doctoral Thesis prepared between the two cities of Rome and Barcelona. This means that there are many people I’d the opportunity to meet, to work with, to share wonderful and terrible moments, and that I’d like to thank in these pages. First of all, to Dr. Piergiorgio Zuccaro, my “creator”, who always believed in me, supported my job and hardly fought to give me all the possible opportunities to grow up as an investigator; To Dr. -

(19) United States (12) Patent Application Publication (10) Pub
US 20130289061A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2013/0289061 A1 Bhide et al. (43) Pub. Date: Oct. 31, 2013 (54) METHODS AND COMPOSITIONS TO Publication Classi?cation PREVENT ADDICTION (51) Int. Cl. (71) Applicant: The General Hospital Corporation, A61K 31/485 (2006-01) Boston’ MA (Us) A61K 31/4458 (2006.01) (52) U.S. Cl. (72) Inventors: Pradeep G. Bhide; Peabody, MA (US); CPC """"" " A61K31/485 (201301); ‘4161223011? Jmm‘“ Zhu’ Ansm’ MA. (Us); USPC ......... .. 514/282; 514/317; 514/654; 514/618; Thomas J. Spencer; Carhsle; MA (US); 514/279 Joseph Biederman; Brookline; MA (Us) (57) ABSTRACT Disclosed herein is a method of reducing or preventing the development of aversion to a CNS stimulant in a subject (21) App1_ NO_; 13/924,815 comprising; administering a therapeutic amount of the neu rological stimulant and administering an antagonist of the kappa opioid receptor; to thereby reduce or prevent the devel - . opment of aversion to the CNS stimulant in the subject. Also (22) Flled' Jun‘ 24’ 2013 disclosed is a method of reducing or preventing the develop ment of addiction to a CNS stimulant in a subj ect; comprising; _ _ administering the CNS stimulant and administering a mu Related U‘s‘ Apphcatlon Data opioid receptor antagonist to thereby reduce or prevent the (63) Continuation of application NO 13/389,959, ?led on development of addiction to the CNS stimulant in the subject. Apt 27’ 2012’ ?led as application NO_ PCT/US2010/ Also disclosed are pharmaceutical compositions comprising 045486 on Aug' 13 2010' a central nervous system stimulant and an opioid receptor ’ antagonist. -

Poisons Act.Fm
LAWS OF BRUNEI CHAPTER 114 POISONS Enactment No. 13 of 1956 Amended by S 103/1958 Enactment No. 6 of 1967 S 101/1979 1984 Edition, Chapter 114 Amended by S 16/1996 S 28/2001 GN 273/2002 REVISED EDITION 2015 B.L.R.O. 1/2015 LAWS OF BRUNEI Poisons CAP. 114 1 LAWS OF BRUNEI REVISED EDITION 2015 CHAPTER 114 POISONS ARRANGEMENT OF SECTIONS Section 1. Citation. 2. Interpretation. 3. Description of poisons. 4. Licensing Officers. 5. General prohibition with respect to importation and sale of poisons. 6. Prohibitions and provisions relating to sale of poisons. 7. Exemptions in respect of medicines. 8. Exemptions in respect of sale. 9. Possession of poisons. 10. Issue of licences. 11. Different kinds of licence. 12. Licences to be numbered and registered. 13. Annual list to be published. 14. Forms of licence. 15. Search and search warrants. 16. Powers of exemption. 17. Penalties. B.L.R.O. 1/2015 LAWS OF BRUNEI 2 CAP. 114 Poisons 18. Jurisdiction. 19. Prosecutions. 20. Prohibition of sale to persons under 18. 21. Rules. SCHEDULE — POISONS LIST ____________________________ LAWS OF BRUNEI Poisons CAP. 114 3 POISONS ACT An Act to regulate the importation, possession, manufacture, compounding, storage, transport and sale of poisons Commencement: 1st January 1983 [S 61/1957] Citation. 1. This Act may be cited as the Poisons Act. Interpretation. 2. In this Act, unless the context otherwise requires — “dentist” means a dentist licensed under the Medical Practitioners and Dentists Act (Chapter 112) and includes a Government dentist; “export”, in relation -
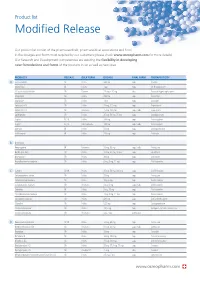
Modified Release
Product list Modified Release Our product list consist of the pharmaceuticals, pharmaceutical associations and food in the dosages and forms most required by our customers (please check www.osmopharm.com for more details). Our Research and Development competencies are assuring the flexibility in developing new formulations and forms of the products in list as well as new ones. PRODUCTS RELEASE BULK FORM DOSAGE FINAL FORM THERAPEUTICITY A Acetazolamide SR Pellets 500 mg caps Diuretic Alfacalcidol IR Pellets 1 μg caps Vit D supplement Alfuzosin Hydrochloride SR Powder 2.5 mg – 10 mg tabs Prostatic Hypertrophy agent Allopurinol SR Pellets 300 mg caps Antiurolytic Alprazolam SR Pellets 1 mg caps Anxiolytic Ambroxol HCI SR Pellets 75 mg, 120 mg caps Expectorant Ambroxol HCI SR Resinates 75 mg, 120 mg caps / tabs Expectorant Amitriptyline SR Pellets 25 mg, 50 mg, 75 mg caps Antidepressant Aspirin EC, IR Pellets 100 mg caps Anticoagulant Aspirin EC, IR Microcapsules 100 mg caps / tabs Anticoagulant Atenolol IR Pellets 50 mg caps Antihypertensive Azithromycin IR Pellets 250 mg caps Antibiotic B B-complex Benproperine IR Resinates 25 mg, 50 mg caps / tabs Antitussive Betahistine 2HCI SR Pellets 12 mg, 24 mg, 48 mg caps Vasodilator Bromopride* SR Pellets 20 mg caps Antiemetic Brompheniramine maleate SR Pellets 6 mg, 8 mg, 12 mg caps Antihistaminic C Caffeine SR, IR Pellets 25 mg, 50 mg, 300 mg caps CNS Stimulant Carbetapentane citrate SR Pellets 75 mg caps Antitussive Carbinoxamine maleate SR Pellets 4mg, 6 mg caps Antihistaminic Carbinoxamine -

Pdf 36.06 Kb
PROJECT REVIEW “Synthesis and Characterisation of Metabolites for the Integration in a Comprehensive Screening Procedure utilizing LC-MS/MS” W. Schänzer, M. Parr (German Sport University, Cologne, Germany) In the fight against doping the laboratories are confronted with an increasing number of substances to screen on. Therefore new methods have to be implemented by the laboratories. To keep the costs for doping control analysis acceptable, to ensure rapid reporting times and to lower the amount of urine needed to screen for all substances, a comprehensive screening for different classes of substances is desirable. Following the 2005 application WADA has granted a pilot project to check for the applicability of direct LC-MS/MS measurement of sulfoconjugates of heavy volatile stimulants. As most of the beta-2 agonists and heavy volatilestimulants are conjugated to sulfuric acid in humans an extension of the method to other compounds is desirable. As reference substances of the conjugates are barely available, during the pilot project the sulfoconjugates of p-Hydroxyamphetamine, p-Hydroxymetamphetamine (Pholedrine), p- Hydroxyephedrine, p-Hydroxynorephedrine, Etilefrine and Etamivan were synthesized by coupling the aglycons to sulfuric acid by reaction with sulfur trioxide pyridine complex. The objective of the continuation is to extend the combined screening procedure for diuretics and heavy volatile stimulants developed in the pilot project to other compounds. Thus, studies on the metabolism have to be reviewed and relevant metabolites have to be synthesised. The structures of all relevant products will be confirmed by nuclear magnetic resonance. For the integration in a comprehensive screening procedure the analytes will be characterised by means of LC-MS/MS. -

The Use of Stems in the Selection of International Nonproprietary Names (INN) for Pharmaceutical Substances
WHO/PSM/QSM/2006.3 The use of stems in the selection of International Nonproprietary Names (INN) for pharmaceutical substances 2006 Programme on International Nonproprietary Names (INN) Quality Assurance and Safety: Medicines Medicines Policy and Standards The use of stems in the selection of International Nonproprietary Names (INN) for pharmaceutical substances FORMER DOCUMENT NUMBER: WHO/PHARM S/NOM 15 © World Health Organization 2006 All rights reserved. Publications of the World Health Organization can be obtained from WHO Press, World Health Organization, 20 Avenue Appia, 1211 Geneva 27, Switzerland (tel.: +41 22 791 3264; fax: +41 22 791 4857; e-mail: [email protected]). Requests for permission to reproduce or translate WHO publications – whether for sale or for noncommercial distribution – should be addressed to WHO Press, at the above address (fax: +41 22 791 4806; e-mail: [email protected]). The designations employed and the presentation of the material in this publication do not imply the expression of any opinion whatsoever on the part of the World Health Organization concerning the legal status of any country, territory, city or area or of its authorities, or concerning the delimitation of its frontiers or boundaries. Dotted lines on maps represent approximate border lines for which there may not yet be full agreement. The mention of specific companies or of certain manufacturers’ products does not imply that they are endorsed or recommended by the World Health Organization in preference to others of a similar nature that are not mentioned. Errors and omissions excepted, the names of proprietary products are distinguished by initial capital letters. -

KI-311UR-IMM Vab Methamphetamine Urine HEIA
HANDLE ALL URINE SPECIMENS AS IF THEY ARE POTENTIALLY INFECTIOUS. Catalog Number: 311UR-0025 25 mL Kit Catalog Number: 311UR-0100 100 mL Kit ASSAY PROCEDURE Catalog Number: 311UR-0500 500 mL Kit Analyzers capable of maintaining a constant temperature, pipetting samples and Catalog Number: 311UR-0060W 60 mL Kit reagents, mixing reagents, timing the reaction accurately and measuring enzymatic Intended Use: rates at 340 nm can be used to perform the assay. The assay has been validated for The Immunalysis Methamphetamine Urine Enzyme Immunoassay is a homogeneous use with the Olympus AU400e, the following Beckman Coulter analyzers: AU480, enzyme immunoassay with a dual cutoff of 500ng/mL and 1000ng/mL. The assay is AU680, AU2700, AU5400 and AU5800. intended for use in laboratories for the qualitative and semi-quantitative analysis of Methamphetamine in human urine with automated clinical chemistry analyzers. This assay Refer to the analyzer-specific Application Sheet which contains parameters for use. is calibrated against Methamphetamine. This in-vitro diagnostic device is for prescription QUALITY CONTROL AND CALIBRATION use only. Good laboratory practice suggests the use of control specimens to ensure assay The semi-quantitative mode is for purposes of enabling laboratories to determine an performance. Control results must fall within established ranges determined by your appropriate dilution of the specimen for confirmation by a confirmatory method such as laboratory. QC materials should be used in accordance with local, state and/or federal Gas Chromatography/ Mass Spectrometry (GC-MS) or permitting laboratories to establish regulations or accreditation requirements. quality control procedures. For a qualitative analysis use either the 500ng/mL or 1000ng/mL calibrator as a cutoff The Immunalysis Methamphetamine Urine Enzyme Immunoassay Kit provides only level to distinguish “positive” and “negative” specimens. -

NIDA Drug Supply Program Catalog, 25Th Edition
RESEARCH RESOURCES DRUG SUPPLY PROGRAM CATALOG 25TH EDITION MAY 2016 CHEMISTRY AND PHARMACEUTICS BRANCH DIVISION OF THERAPEUTICS AND MEDICAL CONSEQUENCES NATIONAL INSTITUTE ON DRUG ABUSE NATIONAL INSTITUTES OF HEALTH DEPARTMENT OF HEALTH AND HUMAN SERVICES 6001 EXECUTIVE BOULEVARD ROCKVILLE, MARYLAND 20852 160524 On the cover: CPK rendering of nalfurafine. TABLE OF CONTENTS A. Introduction ................................................................................................1 B. NIDA Drug Supply Program (DSP) Ordering Guidelines ..........................3 C. Drug Request Checklist .............................................................................8 D. Sample DEA Order Form 222 ....................................................................9 E. Supply & Analysis of Standard Solutions of Δ9-THC ..............................10 F. Alternate Sources for Peptides ...............................................................11 G. Instructions for Analytical Services .........................................................12 H. X-Ray Diffraction Analysis of Compounds .............................................13 I. Nicotine Research Cigarettes Drug Supply Program .............................16 J. Ordering Guidelines for Nicotine Research Cigarettes (NRCs)..............18 K. Ordering Guidelines for Marijuana and Marijuana Cigarettes ................21 L. Important Addresses, Telephone & Fax Numbers ..................................24 M. Available Drugs, Compounds, and Dosage Forms ..............................25 -

Marrakesh Agreement Establishing the World Trade Organization
No. 31874 Multilateral Marrakesh Agreement establishing the World Trade Organ ization (with final act, annexes and protocol). Concluded at Marrakesh on 15 April 1994 Authentic texts: English, French and Spanish. Registered by the Director-General of the World Trade Organization, acting on behalf of the Parties, on 1 June 1995. Multilat ral Accord de Marrakech instituant l©Organisation mondiale du commerce (avec acte final, annexes et protocole). Conclu Marrakech le 15 avril 1994 Textes authentiques : anglais, français et espagnol. Enregistré par le Directeur général de l'Organisation mondiale du com merce, agissant au nom des Parties, le 1er juin 1995. Vol. 1867, 1-31874 4_________United Nations — Treaty Series • Nations Unies — Recueil des Traités 1995 Table of contents Table des matières Indice [Volume 1867] FINAL ACT EMBODYING THE RESULTS OF THE URUGUAY ROUND OF MULTILATERAL TRADE NEGOTIATIONS ACTE FINAL REPRENANT LES RESULTATS DES NEGOCIATIONS COMMERCIALES MULTILATERALES DU CYCLE D©URUGUAY ACTA FINAL EN QUE SE INCORPOR N LOS RESULTADOS DE LA RONDA URUGUAY DE NEGOCIACIONES COMERCIALES MULTILATERALES SIGNATURES - SIGNATURES - FIRMAS MINISTERIAL DECISIONS, DECLARATIONS AND UNDERSTANDING DECISIONS, DECLARATIONS ET MEMORANDUM D©ACCORD MINISTERIELS DECISIONES, DECLARACIONES Y ENTEND MIENTO MINISTERIALES MARRAKESH AGREEMENT ESTABLISHING THE WORLD TRADE ORGANIZATION ACCORD DE MARRAKECH INSTITUANT L©ORGANISATION MONDIALE DU COMMERCE ACUERDO DE MARRAKECH POR EL QUE SE ESTABLECE LA ORGANIZACI N MUND1AL DEL COMERCIO ANNEX 1 ANNEXE 1 ANEXO 1 ANNEX -

2012 Anti-Doping Testing Figures Report Page 1 of 149
2012 Anti-Doping Testing Figures Report ____________________________________________________________________________________ Table of Contents Section 1: 2012 Figures by Laboratory Table A1: Total Samples Analyzed (All Sports) Table A2: Comparison of Years 2008 to 2012 - Olympic and Non-Olympic Figures Table B1: Summary - Samples Analyzed per Laboratory Table B2: Summary - Samples Analyzed per Laboratory (reported in ADAMS) Table B3: Summary - Samples Analyzed per Laboratory (not reported in ADAMS) Table B4: Total Samples Analyzed per Laboratory IC and OOC (reported in ADAMS) Table B5: Total Samples Analyzed per Laboratory IC and OOC (not reported in ADAMS) Table B6: Total Samples Analyzed per Laboratory in Olympic Sports Table B7: Total Samples Analyzed per Laboratory in non-Olympic Sports Table C1: GC/C/IRMS and EPO Tests Conducted per Laboratory Table C2: hGH, Transfusion and HBOCs Tests Conducted per Laboratory Section 2: 2012 Figures by Substance Table D1: Summary - Substances Identified in Each Drug Class in ADAMS (All Sports) Table D2: Substances Identified in Each Drug Class in ADAMS (All Sports) Table D3: Total Laboratory AAFs and ATFs per Drug Class in ADAMS (All Sports) Section 3: 2012 Figures by Sport Table E1: Total Samples Analyzed in Olympic Sports Table E2: Total Samples Analyzed in IOC Recognized Sports Table E3: Total Samples Analyzed in AIMS Sports Table E4: Total Samples Analyzed in Other Sports Table E5: GC/C/IRMS and EPO Tests Conducted per Olympic Sport Table E6: GC/C/IRMS and EPO Tests Conducted per IOC Recognized -

Federal Register / Vol. 60, No. 80 / Wednesday, April 26, 1995 / Notices DIX to the HTSUS—Continued
20558 Federal Register / Vol. 60, No. 80 / Wednesday, April 26, 1995 / Notices DEPARMENT OF THE TREASURY Services, U.S. Customs Service, 1301 TABLE 1.ÐPHARMACEUTICAL APPEN- Constitution Avenue NW, Washington, DIX TO THE HTSUSÐContinued Customs Service D.C. 20229 at (202) 927±1060. CAS No. Pharmaceutical [T.D. 95±33] Dated: April 14, 1995. 52±78±8 ..................... NORETHANDROLONE. A. W. Tennant, 52±86±8 ..................... HALOPERIDOL. Pharmaceutical Tables 1 and 3 of the Director, Office of Laboratories and Scientific 52±88±0 ..................... ATROPINE METHONITRATE. HTSUS 52±90±4 ..................... CYSTEINE. Services. 53±03±2 ..................... PREDNISONE. 53±06±5 ..................... CORTISONE. AGENCY: Customs Service, Department TABLE 1.ÐPHARMACEUTICAL 53±10±1 ..................... HYDROXYDIONE SODIUM SUCCI- of the Treasury. NATE. APPENDIX TO THE HTSUS 53±16±7 ..................... ESTRONE. ACTION: Listing of the products found in 53±18±9 ..................... BIETASERPINE. Table 1 and Table 3 of the CAS No. Pharmaceutical 53±19±0 ..................... MITOTANE. 53±31±6 ..................... MEDIBAZINE. Pharmaceutical Appendix to the N/A ............................. ACTAGARDIN. 53±33±8 ..................... PARAMETHASONE. Harmonized Tariff Schedule of the N/A ............................. ARDACIN. 53±34±9 ..................... FLUPREDNISOLONE. N/A ............................. BICIROMAB. 53±39±4 ..................... OXANDROLONE. United States of America in Chemical N/A ............................. CELUCLORAL. 53±43±0 -

Poisons Act 1957 01.01.2013.Pdf
Singapore Statutes Online - 1 - Poisons Rules Page 1 of 125 Poisons Rules 1 Citation 2 Definitions 3 Licences 4 Form of record of sales 5 Wholesale sales 6 (Deleted) 7 (Deleted) 8 Fees 9 Extension of labelling provisions 10 Limitation of section 6 (3) of Act to certain substances 11 Extension of section 6 (3) to section 8 of Act, sales and supply by commercial sample, and relaxation of this subsection 12 Relaxation of section 7 (3) of Act in case of certain medicines 13 Exemption from provisions relating solely to substances in First Schedule 14 Complete exemption of certain articles and poisons 14A Exemption for retail sale of poisons by pharmacists 14B (Deleted) 14C (Deleted) 15 Additional restrictions on sale of certain poisons 16 Coded prescription prohibited 17 Restrictions on sale of codeine cough preparations by pharmacists 18 Restriction of sale of strychnine 19 Manner of labelling containers 20 Labelling of name of poison 21 Labelling of particulars as to proportion of poison 22 Indication of character of poison 23 Special cautions in case of certain substances 24 Relaxation of certain labelling requirements 25 Form of containers 26 Storage of poisons 27 Transport of poisons 28 Special provisions with respect to transport of certain poisons 29 Supply of medicines to outpatients from certain hospitals, etc. 30 Supply of medicines for use in hospitals, etc. 31 Storage of poisons in institutions 32 Manufacture of pharmaceutical preparations 33 Colouring of poisons 34 Preservation of records 35 Penalties FIRST SCHEDULE Substances falling within the Poisons List to which special restrictions under rule 10 apply http://statutes.agc.gov.sg/aol/search/display/printView.w3p;page=0;query=Id%3A%2..