Bench Scale Production of Ammonium Potassium Polyphosphate Charles Arthur Hodge Iowa State University
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Evaluation of Liquid Ammonium Polyphosphate As a Carrier of Iron and Zinc
EVALUATION OF LIQUID AMMONIUM POLYPHOSPHATE AS A CARRIER OF IRON AND ZINC by ; V<P RATMUNDO RALLAN GANIRON B. S. A., University of the Philippines, 1961 A MASTER'S THESIS submitted in partial fulfillment of the requirement for the degree MASTER OF SCIENCE Department of Agronomy KANSAS STATE UNIVERSITY Manhattan, Kansas 1966 Approved by: Jor Professor LP ZGGf it Tj( £/<?7 TABLE OF CONTENTS INTRODUCTION 1 REVIEW OF LITERATURE 3 Iron . 3 Zinc * MATERIALS AND METHODS 13 Fertiliser Materials 13 Field Experiments H Soil Sampling 20 Laaf Sampling 22 Chemical Analysis 22 (a) Zinc 22 (b) Iron 24 (c) Phosphorua 25 (d) Potassium •« 27 (a) Organic Matter 27 (f) pH 27 (g) Protain 27 RESULTS AND DISCUSSIONS 26 SUMMARY AND CONCLUSIONS 4$ ACKNOWLEDGMENT. 51 LITERATURE CITED 52 INTRODUCTION Increasing attention has baan focused to the need of fertilising with micronutrient elements to achieve maximum production and optimum quality of farm crops. The supply of these elements in the soil has become limited in some areas as a result of the introduction of high yielding varieties, better and more intensive cropping practices, and increased use of high analysis fertilisers. In some cases, the ob- served deficiencies of these micronutrients have been man- made due to addition of interfering elements for other pur- poses. For instance there is the copper induced iron chlorosis, copper being introduced as agricultural sprays and the phosphorus induced iron and zinc deficiencies, phos- phorus being introduced to meet the phosphorus requirement of farm crops. How to minimise such interactions and the problems of keeping these micronutrient elements available to the plants when applied to the soil have been the sub- ject of intensive researches. -

Potassium Cyanide Broth Base W/O KCN M936
Potassium Cyanide Broth Base w/o KCN M936 Potassium Cyanide Broth Base with KCN supplementation is used for the differentiation of the members of Enterobacteriaceae on the basis of potassium cyanide tolerance. Composition** Ingredients Gms / Litre Proteose peptone 3.000 Disodium phosphate 5.640 Monopotassium phosphate 0.225 Sodium chloride 5.000 Final pH ( at 25°C) 7.6±0.2 **Formula adjusted, standardized to suit performance parameters Directions Suspend 13.86 grams in 1000 ml distilled water. Heat if necessary to dissolve the medium completely. Dispense in 100 ml amounts and sterilize by autoclaving at 15 lbs pressure (121°C) for 15 minutes. Cool to room temperature and aseptically add sterile 1.5 ml of 0.5% potassium cyanide solution to each 100 ml of basal medium. Mix thoroughly and dispense in 1 ml amounts.Caution : Being fatally toxic extreme care should be taken while handling potassium cyanide solution. Never mouth- pipette potassium cyanide solution. Principle And Interpretation One of the many tests employed for the identification of bacteria includes the ability of an organism to grow in the presence of cyanide (1). Potassium Cyanide Broth Base is used for the differentiation of members of Enterobacteriaceae on the basis of Potassium Cyanide tolerance. Potassium Cyanide Broth Base was originally formulated by Moeller (2) and Kauffman and Moeller (3). This medium was later modified by Edwards and Ewing (4) and Edwards and Fife (5). Proteose peptone provides nitrogenous compounds, sulphur, trace elements essential for growth. Phosphates buffer the medium. Sodium chloride maintains osmotic equilibrium. Potassium cyanide inhibits many bacteria including Salmonella , Shigella and Escherichia , while members of the Klebsiella , Citrobacter , and Proteus groups grow well. -

Effects of Different Sources of Fertilizer Nitrogen on Growth and Nutrition of Western Hemlock Seedlings
Effects of Different Sources U.S. Department of Agriculture Forest Service Pacific Northwest Forest of FertiIizer Nitrogen and Range Experiment Station Research Paper PNW-267 on Growth and Nutrition oJ February 1980 Western Hemlock Seedlings ---. --_. ------------------------ , I _J Authors M. A. RADWAN is Principal Plant Physiologist and DEAN S. DeBELL is Principal Silviculturist with the Forest Service, u.S. Department of Agriculture, Pacific Northwest Forest and Range Experiment Station, Forestry Sciences Laboratory, Olympia, Washington. En gl ish Equivalents 1 liter 0.2642 gallon 1 kilogram = 2.2046 pound 1 gram = 0.0353 ounce 1 centimeter = 0.3937 inch 1 kilogram per hectare 1.1206 pounds per acre (9/50C) + 32 = of EFFECTS OF DIFFERENT SOURCES OF FERTILIZER NITROGEN ON GROWTH AND NUTRITION OF WESTERN HEMLOCK Reference Abstract Radwan, M. A. , and Dean S. DeBell. 1980. Effects of different sources of fertilizer nitrogen on growth and nutrition of western hemlock seedlings. USDA For. Servo Res. Pap. PNW-267, 15 p. Pacific Northwest Forest and Range Experiment Station, Portland, Oregon. Twelve different nitrogen (N) fertilizer treatments were tested on potted western hemlock (Tsuga heterophylla (Raf. ) Sarg.) seedlings. Fertilizers affected soil N and pH, and growth and foliar chemical com position of seedlings. Ura plus N-Serve and sulfur-coated urea appear more promising for promoting growth than other fertilizers tested. Results, however, do not explain reported variability in response of hemlock stands to N fertilization. Keywords: Nitrogen fertilizer response, seedling growth, western hemlock, Tsuga heterophylla. RESEARCH SUMMARY Research Paper PNW-267 1980 The following fertilization treatments were applied in the spring to potted, 4-year-old western hemlock (Tsuga heterophylla (Raf. -

Brochure-Product-Range.Pdf
PRODUCT RANGE 2015 edition ANSI Standard 60 NSF® CERTIFIED HALAL M ISLAMIC FOOD AND NUTRITION ® COUNCIL OF AMERICA Rue Joseph Wauters, 144 ISO 9001:2008 (Quality) / OHSAS 18001:2007 (Health/ B-4480 Engis Safety) / ISO 14001:2004 (Environment) / ISO 22000:2005 www.globulebleu.com (Food Safety) / FSSC 22000:2013 (Food Safety). Tel. +32 (0) 4 273 93 58 Our food grade phosphates are allergen free, GMO free, Fax. +32 (0) 4 275 68 36 BSE/TSE free. www.prayon.com mail. [email protected] Design by www.prayon.com PRODUCT RANGE | 11 TABLE OF CONTENTS HORTICULTURE APPLICATIONS HORTIPRAY® RANGE FOR HORTICULTURE* FOOD AND INDUSTRIAL APPLICATIONS PRODUCT NAME Bulk density P O pH N-NH Made 2 5 4 MONOAMMONIUM PHOSPHATE - NH4H2PO4 in 3 3 % 1% % Sodium orthophosphates ................................................................................... 03 g/cm lbs/ft indicative indicative indicative Water-soluble fertilisers. Sodium pyrophosphates .................................................................................... 04 HORTIPRAY® MAP Horticultural Grade 0.9 56 61 4.5 12 Sodium tripolyphosphates ................................................................................. 05 HORTIPRAY® MAP 12.60 Horticultural Grade 0.9 56 60 5 12.1 Water-soluble fertilisers; Sodium polyphosphates ..................................................................................... 06 HORTIPRAY® MAP anticalc Horticultural Grade 0.9 56 61 4.5 12 preventive action against clogging. Potassium orthophosphates ............................................................................. -

United States Patent (19) 11 Patent Number: 5,788,915 Blount (45) Date of Patent: Aug
IIIUSOO5788915A United States Patent (19) 11 Patent Number: 5,788,915 Blount (45) Date of Patent: Aug. 4, 1998 54 FLAME RETARDANT COMPOSITIONS 57 ABSTRACT UTILIZING PARTIALLY HYDROLYZED AMNO CONDENSATON COMPOUNDS Flame retardant compositions of this invention are produced by incorporating a partially index (LOI) hydrolyzed amino 76 Inventor: David H. Blount, 6728 Del Cerro condensation composition in a more flammable organic Blvd., San Diego, Calif. 92120 material. The partially hydrolyzed amino condensation com pounds are produced by heating urea or heating urea with (21) Appl. No.: 801,776 other nitrogen containing compounds that will condensate with or react with isocyanic acid and/or cyanic acid thereby 22 Filed: Feb. 14, 1997 producing an amino condensation compound which is then Related U.S. Application Data partially hydrolysis is done by reacting it with a limited amount of water. The partially hydrolyzed amino conden 62) Division of Ser. No. 723,779, Sep. 30, 1996. sation compounds may be used alone or may be mixed with or reacted with carbonization auxiliaries, aldehydes and (51) Int. Cl. ........................ C09K 21A00; C08G 12/12 fillers to produce a partially hydrolyzed amino condensation 52 U.S. Cl. ........................... 252/609; 528/259; 252/601 composition which is incorporated in more flammable 58) Field of Search ..................................... 252/609, 601: organic compositions such as polyurethanes, polyester 528/259 resins. epoxy resins, vinyl resins and other resins. The partially hydrolyzed amino condensation salts of 56 References Cited phosphorus, boron or sulfur containing compounds and the U.S. PATENT DOCUMENTS partially hydrolyzed amino condensation-aldehyde resins 3,900,665 8/1975 Weil ....................................... -

Phase-Matched Metamaterials for Second-Harmonic Generation
Anna Vesala PHASE-MATCHED METAMATERIALS FOR SECOND-HARMONIC GENERATION Faculty of Engineering and Natural Sciences (ENS) Bachelor of Science Thesis April 2020 i ABSTRACT Anna Vesala: Phase-matched Metamaterials for Second-harmonic Generation Bachelor of Science Thesis Tampere University Bachelors Degree Programme in Science and Engineering Major: Physics Examiners: Dr. Mikko Huttunen and M.Sc. Timo Stolt April 2020 Metamaterials exhibit unconventional electromagnetic properties that cannot be found in nature, such as negative index of refraction or strong optical activity. Moreover, they show promise for enabling nanoscale nonlinear optics. Current nonlinear optical interactions of practical use rely on phase matching combined with long propagation lengths, which are not compatible with the size requirements of miniaturized systems. In order to be able to improve the realizable conversion efficiencies of nonlinear processes and discover novel functionalities at the nanoscale, new kinds of nonlinear metamaterials need to be investigated. By utilizing local-field enhancements and the phase engineering of localized surface plas- mon resonances, it is possible to construct metamaterials which generate nonlinear frequencies into the direction where the fundamental light came from. In this Thesis, we demonstrate how phase matching is achieved in nanoscale nonlinear materials. Especially, we fabricate three- dimensional plasmonic metamaterial devices that were phase matched for back-propagating sec- ond harmonic-generation. Our samples consist of one to five metasurfaces stacked on top of each other and the aim was to observe how the intensity of the second-harmonic field varies with the number of metasurfaces stacked in a backward phase-matched metamaterial. The results show that the second harmonic signal depends quadratically on the number of metasurfaces, which confirms that the sample was successfully phase-matched by controlling the dimensions of the nanoparticles and the separation between the metasurfaces. -
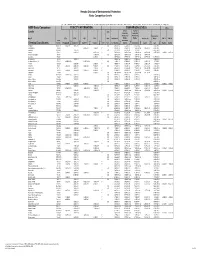
Basic Comparison Levels
Nevada Division of Environmental Protection Basic Comparison Levels Key: I=IRIS; P= PPRTV; N=NCEA; H=HEAST; A=ATSDR; O=Other Documents; CA=CalEPA S=Surrogate X=Appendix PPRTV E=Based on TEF scheme r=Route Extra Key: C = Cancer endpoint; N = Noncancer endpoint; sat = Saturation Limit; max = Ceiling Limit NDEP Basic Comparison TOXICITY INFORMATION COMPARISON LEVELS LBCLs Indoor Outdoor Levels Skin Industrial/ Industrial/ Commercial Commercial Residential May-17 SFo RfDo IUR RfCi Abs. Residential Worker Worker Ambient Air Water DAF 1 DAF 20 CAS w/o Dermal Chemical Constituents Number 1/(mg/kg-d) (mg/kg-d) (ug/m3)-1 (mg/m3) VOCc Soils Soil (mg/kg) (mg/kg) Soil (mg/kg) (µg/m3) (µg/l) (mg/kg) (mg/kg) Key Key key Key Key Key Key Key Key Acephate 30560-19-1 8.70E-03 I 4.00E-03 I 0.10 5.59E+01 C 7.52E+02 C 2.95E+02 C 7.73E+00 C Acetaldehyde 75-07-0 2.20E-06 I 9.00E-03 I V 1.23E+01 C 5.35E+01 C 1.00E+05 max 1.28E+00 C 2.55E+00 C Acetochlor 34256-82-1 2.00E-02 I 0.10 1.23E+03 N 4.67E+04 N 1.83E+04 N 6.67E+02 N Acetone 67-64-1 9.00E-01 I 3.10E+01 A V 7.04E+04 N 1.00E+05 max 1.00E+05 max 3.23E+04 N 2.05E+04 N 8.00E-01 1.60E+01 Acetone Cyanohydrin 75-86-5 2.00E-03 X 0.10 1.00E+05 max 1.00E+05 max 1.00E+05 max 2.09E+00 N Acetonitrile 75-05-8 6.00E-02 I V 1.00E+05 max 3.75E+03 N 1.00E+05 max 6.26E+01 N 1.25E+02 N Acetophenone 98-86-2 1.00E-01 I V 2.52E+03 sat 2.52E+03 sat 2.52E+03 sat 3.34E+03 N Acetylaminofluorene, 2- 53-96-3 3.80E+00 CA 1.30E-03 CA 0.10 1.28E-01 C 1.72E+00 C 6.75E-01 C 2.16E-03 C 1.77E-02 C Acrolein 107-02-8 5.00E-04 I 2.00E-05 I V -

11-37-0 Liquid Ammonium Polyphosphate Guaranteed Analysis Total Nitrogen (N)
Product Data Sheet 11-37-0 LIQUID AMMONIUM POLYPHOSPHATE GUARANTEED ANALYSIS TOTAL NITROGEN (N) ................................................................................................................................ 11.00% 11.00% Ammoniacal Nitrogen AVAILABLE PHOSPHATE (P2O5) .................................................................................................................37.00% Molybdenum (Mo).........................................................................................................................................0.001% Derived from Ammonium Phosphate. KEEP OUT OF REACH OF CHILDREN tanks and certain fiberglass tanks. Aluminum tanks WARNING can also be used. However, aluminum tanks are not CAUSES SKIN IRRITATION. CAUSES EYE recommended for prolonged storage. IRRITATION. MAY CAUSE RESPIRATORY 2. Pumps: Any pump recommended for use with liquid IRRITATION. fertilizers. Remember the basic rule of "no brass." PRECAUTIONARY STATEMENTS: Avoid breathing fumes / gas / mist / vapors / spray. Wash thoroughly after handling. Use only USES outdoors or in a well-ventilated area. Call a poison center / doctor if 1. Is an excellent source of water soluble, fast acting you feel unwell. Specific treatment see First Aid section on this label. ammonium phosphate with the most desirable ratio of PERSONAL PROTECTIVE EQUIPMENT: Wear protective N:P2O5. gloves / protective clothing / eye protection / face protection. Take off 2. An ideal source of phosphate and nitrogen in complete contaminated clothing and wash before reuse. liquid mixes. FIRST AID: IF ON SKIN Wash with plenty of water. If skin irritation 3. For specific application rates follow the recommendation occurs, get medical advice / attention. IF INHALED Remove person of a qualified individual or institution, such as, but to fresh air and keep comfortable for breathing. IF IN EYES Rinse not limited to, a certified crop advisor, agronomist, cautiously with water for several minutes. Remove contact lenses, if university crop extension publication, or apply present and easy to do. -
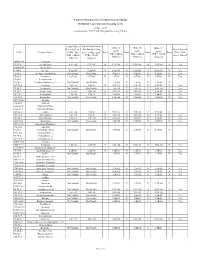
(GWSL) Ug/L TCR=1.0E-05 THQ=0.2 Sub-Slab and Exterior
North Carolina Department of Environmental Quality Residential Vapor Intrusion Screening Levels October 2017 (based on June 2017 USEPA Regional Screening Tables) Ground Water Sub-slab and Exterior IASL (A) IASL (B) IASL (C)3 Screening Level Soil Gas Screening 3 3 3 Does Chemical 3 ug/m ug/m ug/m CAS # Chemical Name: (GWSL) ug/L Level (SGSL) ug/m Basis Basis Basis Basis Have a Non- TCR=1.0E-06 TCR=1.0E-05 TCR=1.0E-04 4 TCR=1.0E-05 TCR=1.0E-05 Cancer Effect? THQ=0.2 THQ=0.2 THQ=0.2 THQ=0.2 THQ=0.2 30560-19-1 Acephate 75-07-0 Acetaldehyde 6.9E+02 6.3E+01 N 1.3E+00 C 1.9E+00 N 1.9E+00 N Yes 34256-82-1 Acetochlor 67-64-1 Acetone 4.5E+06 2.2E+05 N 6.5E+03 N 6.5E+03 N 6.5E+03 N Yes 75-86-5 Acetone Cyanohydrin Not Volatile Not Volatile 4.2E-01 N 4.2E-01 N 4.2E-01 N Yes 75-05-8 Acetonitrile 8.9E+03 4.2E+02 N 1.3E+01 N 1.3E+01 N 1.3E+01 N Yes 98-86-2 Acetophenone 53-96-3 Acetylaminofluorene, 2- Not Volatile Not Volatile 2.2E-03 C 2.2E-02 C 2.2E-01 C 107-02-8 Acrolein 8.4E-01 1.4E-01 N 4.2E-03 N 4.2E-03 N 4.2E-03 N Yes 79-06-1 Acrylamide Not Volatile Not Volatile 1.0E-02 C 1.0E-01 C 1.0E+00 C Yes 79-10-7 Acrylic Acid 1.4E+04 7.0E+00 N 2.1E-01 N 2.1E-01 N 2.1E-01 N Yes 107-13-1 Acrylonitrile 7.3E+01 1.4E+01 C 4.1E-02 C 4.1E-01 C 4.2E-01 N Yes 111-69-3 Adiponitrile Not Volatile Not Volatile 1.3E+00 N 1.3E+00 N 1.3E+00 N Yes 15972-60-8 Alachlor 116-06-3 Aldicarb 1646-88-4 Aldicarb Sulfone 1646-87-3 Aldicarb sulfoxide 309-00-2 Aldrin 3.2E+00 1.9E-01 C 5.7E-04 C 5.7E-03 C 5.7E-02 C 107-18-6 Allyl Alcohol 1.0E+02 7.0E-01 N 2.1E-02 N 2.1E-02 N 2.1E-02 -

Engineering Alcohol Tolerance in Yeast
Engineering alcohol tolerance in yeast The MIT Faculty has made this article openly available. Please share how this access benefits you. Your story matters. Citation Lam, F. H., A. Ghaderi, G. R. Fink, and G. Stephanopoulos. “Engineering Alcohol Tolerance in Yeast.” Science 346, no. 6205 (October 2, 2014): 71–75. As Published http://dx.doi.org/10.1126/science.1257859 Publisher American Association for the Advancement of Science (AAAS) Version Author's final manuscript Citable link http://hdl.handle.net/1721.1/99498 Terms of Use Article is made available in accordance with the publisher's policy and may be subject to US copyright law. Please refer to the publisher's site for terms of use. Title: Engineering alcohol tolerance in yeast Authors: Felix H. Lam1,2, Adel Ghaderi1, Gerald R. Fink2*, Gregory Stephanopoulos1* Affiliations: 1 Department of Chemical Engineering, Massachusetts Institute of Technology, Cambridge, Massachusetts, USA 2 Whitehead Institute for Biomedical Research, Cambridge, Massachusetts, USA * Correspondence to: G.R.F. ([email protected]) or G.S. ([email protected]) Abstract: Ethanol toxicity in yeast Saccharomyces cerevisiae limits titer and productivity in the industrial production of transportation bioethanol. We show that strengthening the opposing potassium and proton electrochemical membrane gradients is a mechanism that enhances general resistance to multiple alcohols. Elevation of extracellular potassium and pH physically bolster these gradients, increasing tolerance to higher alcohols and ethanol fermentation in commercial and laboratory strains (including a xylose-fermenting strain) under industrial-like conditions. Production per cell remains largely unchanged with improvements deriving from heightened population viability. Likewise, up-regulation of the potassium and proton pumps in the laboratory strain enhances performance to levels exceeding industrial strains. -

Contents / Iii
FCC 11 Contents / iii Contents 1 I; ~ith ~er PREFACE v i I PEOPLE xi NEW AND CHANGED..................................................................................................................................................... xiii GENERAL PROVISIONS AND REQUIREMENTS APPLYING TO SPECIFICATIONS, TESTS, AND ASSAYS OF THE FOOD CHEMICALS CODEX .. MONOGRAPHS.................................................................................................................................................................. 9 should IDENTITY STANDARDS.............................................................................................................................................. 1269 GENERAL TESTS AND ASSAyS................................................................................................................................... 1275 Appendix I: Apparatus for Tests and Assays.............................................................................................................. 1279 Appendix II: Physical Tests and Determinations........................................................................................................ 1282 A. Chromatography......................... 1282 B. Physicochemical Properties......................................................................................................................... 1290 C. Others............................. 1302 Appendix III: Chemical Tests and Determinations.................................................................................................... -

Third Supplement, FCC 11 Index / All-Trans-Lycopene / I-1
Third Supplement, FCC 11 Index / All-trans-Lycopene / I-1 Index Titles of monographs are shown in the boldface type. A 2-Acetylpyridine, 20 Alcohol, 80%, 1524 3-Acetylpyridine, 21 Alcohol, 90%, 1524 Abbreviations, 6, 1726, 1776, 1826 2-Acetylpyrrole, 21 Alcohol, Absolute, 1524 Absolute Alcohol (Reagent), 5, 1725, 2-Acetyl Thiazole, 18 Alcohol, Aldehyde-Free, 1524 1775, 1825 Acetyl Valeryl, 562 Alcohol C-6, 579 Acacia, 556 Acetyl Value, 1400 Alcohol C-8, 863 ªAccuracyº, Defined, 1538 Achilleic Acid, 24 Alcohol C-9, 854 Acesulfame K, 9 Acid (Reagent), 5, 1725, 1775, 1825 Alcohol C-10, 362 Acesulfame Potassium, 9 Acid-Hydrolyzed Milk Protein, 22 Alcohol C-11, 1231 Acetal, 10 Acid-Hydrolyzed Proteins, 22 Alcohol C-12, 681 Acetaldehyde, 10 Acid Calcium Phosphate, 219, 1838 Alcohol C-16, 569 Acetaldehyde Diethyl Acetal, 10 Acid Hydrolysates of Proteins, 22 Alcohol Content of Ethyl Oxyhydrate Acetaldehyde Test Paper, 1535 Acidic Sodium Aluminum Phosphate, Flavor Chemicals (Other than Acetals (Essential Oils and Flavors), 1065 Essential Oils), 1437 1395 Acidified Sodium Chlorite Alcohol, Diluted, 1524 Acetanisole, 11 Solutions, 23 Alcoholic Potassium Hydroxide TS, Acetate C-10, 361 Acidity Determination by Iodometric 1524 Acetate Identification Test, 1321 Method, 1437 Alcoholometric Table, 1644 Aceteugenol, 464 Acid Magnesium Phosphate, 730 Aldehyde C-6, 571 Acetic Acid Furfurylester, 504 Acid Number (Rosins and Related Aldehyde C-7, 561 Acetic Acid, Glacial, 12 Substances), 1418 Aldehyde C-8, 857 Acetic Acid TS, Diluted, 1524 Acid Phosphatase