Selective N-Terminal Acylation of Peptides and Proteins with a Gly-His Tag Sequence
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Reactions of Benzene & Its Derivatives
Organic Lecture Series ReactionsReactions ofof BenzeneBenzene && ItsIts DerivativesDerivatives Chapter 22 1 Organic Lecture Series Reactions of Benzene The most characteristic reaction of aromatic compounds is substitution at a ring carbon: Halogenation: FeCl3 H + Cl2 Cl + HCl Chlorobenzene Nitration: H2 SO4 HNO+ HNO3 2 + H2 O Nitrobenzene 2 Organic Lecture Series Reactions of Benzene Sulfonation: H 2 SO4 HSO+ SO3 3 H Benzenesulfonic acid Alkylation: AlX3 H + RX R + HX An alkylbenzene Acylation: O O AlX H + RCX 3 CR + HX An acylbenzene 3 Organic Lecture Series Carbon-Carbon Bond Formations: R RCl AlCl3 Arenes Alkylbenzenes 4 Organic Lecture Series Electrophilic Aromatic Substitution • Electrophilic aromatic substitution: a reaction in which a hydrogen atom of an aromatic ring is replaced by an electrophile H E + + + E + H • In this section: – several common types of electrophiles – how each is generated – the mechanism by which each replaces hydrogen 5 Organic Lecture Series EAS: General Mechanism • A general mechanism slow, rate + determining H Step 1: H + E+ E El e ctro - Resonance-stabilized phile cation intermediate + H fast Step 2: E + H+ E • Key question: What is the electrophile and how is it generated? 6 Organic Lecture Series + + 7 Organic Lecture Series Chlorination Step 1: formation of a chloronium ion Cl Cl + + - - Cl Cl+ Fe Cl Cl Cl Fe Cl Cl Fe Cl4 Cl Cl Chlorine Ferric chloride A molecular complex An ion pair (a Lewis (a Lewis with a positive charge containing a base) acid) on ch lorine ch loronium ion Step 2: attack of -

Reactions of Aromatic Compounds Just Like an Alkene, Benzene Has Clouds of Electrons Above and Below Its Sigma Bond Framework
Reactions of Aromatic Compounds Just like an alkene, benzene has clouds of electrons above and below its sigma bond framework. Although the electrons are in a stable aromatic system, they are still available for reaction with strong electrophiles. This generates a carbocation which is resonance stabilized (but not aromatic). This cation is called a sigma complex because the electrophile is joined to the benzene ring through a new sigma bond. The sigma complex (also called an arenium ion) is not aromatic since it contains an sp3 carbon (which disrupts the required loop of p orbitals). Ch17 Reactions of Aromatic Compounds (landscape).docx Page1 The loss of aromaticity required to form the sigma complex explains the highly endothermic nature of the first step. (That is why we require strong electrophiles for reaction). The sigma complex wishes to regain its aromaticity, and it may do so by either a reversal of the first step (i.e. regenerate the starting material) or by loss of the proton on the sp3 carbon (leading to a substitution product). When a reaction proceeds this way, it is electrophilic aromatic substitution. There are a wide variety of electrophiles that can be introduced into a benzene ring in this way, and so electrophilic aromatic substitution is a very important method for the synthesis of substituted aromatic compounds. Ch17 Reactions of Aromatic Compounds (landscape).docx Page2 Bromination of Benzene Bromination follows the same general mechanism for the electrophilic aromatic substitution (EAS). Bromine itself is not electrophilic enough to react with benzene. But the addition of a strong Lewis acid (electron pair acceptor), such as FeBr3, catalyses the reaction, and leads to the substitution product. -

Activation of Pyridinium Salts for Electrophilic Acylation: a Method for Conversion of Pyridines Into 3-Acylpyridines
Chemistry of Heterocyclic Compounds, Vol. 40, No. 6, 2004 ACTIVATION OF PYRIDINIUM SALTS FOR ELECTROPHILIC ACYLATION: A METHOD FOR CONVERSION OF PYRIDINES INTO 3-ACYLPYRIDINES A. Klapars1 and E. Vedejs2 Cyanide adducts of N-MOM pyridinium salts react with strong acylating reagents to provide 3-acyl-4- cyano-1,4-dihydropyridines that can be aromatized to 3-acylpyridines using ZnCl2 in refluxing ethanol. Keywords: dihydropyridines, pyridine acylation. Electrophilic aromatic substitution reactions of pyridines are extraordinarily challenging. Instead of C-substitution at the pyridine ring, the electrophile typically forms an adduct with the pyridine nitrogen, which even further deactivates the already electron deficient pyridine ring toward electrophilic substitution. For example, the direct nitration of pyridine may require a reaction temperature of 330°C to provide only a 15% yield of 3-nitropyridine [1]. To the best of our knowledge, no direct, intermolecular C-acylations of pyridines have been reported [2]. This seriously limits the choice of methods for the preparation of the ubiquitous 3-substituted pyridines [3, 4]. In a limited number of cases, the lack of reactivity of pyridines toward electrophiles has been addressed by converting the recalcitrant pyridine into a temporarily activated 1,4-dihydropyridine [5-9]. In contrast to the electron poor parent pyridine, the electron rich 1,4-dihydropyridine features strongly enhanced reactivity toward electrophiles at the 3-position. Several steps are typically required including formation of the dihydropyridine, the subsequent reaction with an electrophile, and rearomatization to the desired 3-substituted pyridine. A similar concept has been ingeniously employed in a one pot nitration of pyridines in the presence of sulfite as the nucleophile that temporarily activates the pyridine, and then acts as a leaving group in an aromatization step [10, 11]. -

1. A. the First Reaction Is a Friedel-Crafts Acylation (FCA), Where the Major Product Is the Para- Isomer (60% Isolated Yield)
1. a. The first reaction is a Friedel-Crafts Acylation (FCA), where the major product is the para- isomer (60% isolated yield). The second reaction is a nitration, where the incoming electrophile (nitronium ion) is directed to the ortho position of the methoxy group. The last reaction is a Wolff-Kishner reduction that converts the acetyl group into an ethyl group. The nitro group does not react under these conditions. OCH OCH OCH OCH3 3 3 3 NO2 NO2 N2H4/KOH CH3COCl/AlCl3 H2SO4/HNO3 0 oC O CH 3 O CH3 CH3 (A) Reaction 1 (B) Reaction 2 (C) Reaction 3 (P) b. The best solvent for the FC-acylation is dichloromethane. Tetrahydrofuran is a fairly strong Lewis base, which would react and deactivate the AlCl3 catalyst. Ethanol would also react with AlCl3 and form alcoholates, which are inactive at FCA catalyst. Dichloromethane is polar enough to dissolve all three compounds but does not form adducts with AlCl3. Thus, aluminum chloride maintains its Lewis acidity. 3+ c. As discussed in lecture, AlCl3*6 H2O is not suitable as catalyst because the Al is not a strong Lewis acid anymore. In addition, larger amounts of water would destroy the acetyl chloride as well (=hydrolysis, CH3COCl + H2O ---- > CH3COOH + HCl). Consequently, the reaction would not proceed in the desired fashion. 3+ OH2 H2O OH2 Al H2O OH2 OH2 d. In order to determine the yield, one has to calculate the number of moles of the reactant and the product. nA = 1.90 mL * 0.996 g/mL/108.14 g/mol = 17.5 mmol nCH3COCl = 2.49 mL * 1.104 g/mL/78.5 g/mol = 35.0 mmol nAlCl3 = 4.67 g/133.5 g/mol = 35.0 mmol Compound (A) is the limiting reagent. -

Heterocyclic Chemistrychemistry
HeterocyclicHeterocyclic ChemistryChemistry Professor J. Stephen Clark Room C4-04 Email: [email protected] 2011 –2012 1 http://www.chem.gla.ac.uk/staff/stephenc/UndergraduateTeaching.html Recommended Reading • Heterocyclic Chemistry – J. A. Joule, K. Mills and G. F. Smith • Heterocyclic Chemistry (Oxford Primer Series) – T. Gilchrist • Aromatic Heterocyclic Chemistry – D. T. Davies 2 Course Summary Introduction • Definition of terms and classification of heterocycles • Functional group chemistry: imines, enamines, acetals, enols, and sulfur-containing groups Intermediates used for the construction of aromatic heterocycles • Synthesis of aromatic heterocycles • Carbon–heteroatom bond formation and choice of oxidation state • Examples of commonly used strategies for heterocycle synthesis Pyridines • General properties, electronic structure • Synthesis of pyridines • Electrophilic substitution of pyridines • Nucleophilic substitution of pyridines • Metallation of pyridines Pyridine derivatives • Structure and reactivity of oxy-pyridines, alkyl pyridines, pyridinium salts, and pyridine N-oxides Quinolines and isoquinolines • General properties and reactivity compared to pyridine • Electrophilic and nucleophilic substitution quinolines and isoquinolines 3 • General methods used for the synthesis of quinolines and isoquinolines Course Summary (cont) Five-membered aromatic heterocycles • General properties, structure and reactivity of pyrroles, furans and thiophenes • Methods and strategies for the synthesis of five-membered heteroaromatics -

Chapter 16 the Chemistry of Benzene and Its Derivatives
Instructor Supplemental Solutions to Problems © 2010 Roberts and Company Publishers Chapter 16 The Chemistry of Benzene and Its Derivatives Solutions to In-Text Problems 16.1 (b) o-Diethylbenzene or 1,2-diethylbenzene (d) 2,4-Dichlorophenol (f) Benzylbenzene or (phenylmethyl)benzene (also commonly called diphenylmethane) 16.2 (b) (d) (f) (h) 16.3 Add about 25 °C per carbon relative to toluene (110.6 C; see text p. 743): (b) propylbenzene: 161 °C (actual: 159 °C) 16.4 The aromatic compound has NMR absorptions with greater chemical shift in each case because of the ring current (Fig. 16.2, text p. 745). (b) The chemical shift of the benzene protons is at considerably greater chemical shift because benzene is aromatic and 1,4-cyclohexadiene is not. 16.6 (b) Among other features, the NMR spectrum of 1-bromo-4-ethylbenzene has a typical ethyl quartet and a typical para-substitution pattern for the ring protons, as shown in Fig. 16.3, text p. 747, whereas the spectrum of (2- bromoethyl)benzene should show a pair of triplets for the methylene protons and a complex pattern for the ring protons. If this isn’t enough to distinguish the two compounds, the integral of the ring protons relative to the integral of the remaining protons is different in the two compounds. 16.7 (b) The IR spectrum indicates the presence of an OH group, and the chemical shift of the broad NMR resonance (d 6.0) suggests that this could be a phenol. The splitting patterns of the d 1.17 and d 2.58 resonances show that the compound also contains an ethyl group, and the splitting pattern of the ring protons shows that the compound is a para-disubstituted benzene derivative. -

Two Different Facile and Efficient Approaches
Spec. Matrices 2019; 7:1–19 Research Article Open Access Kazumasa Nomura* and Paul Terwilliger Green Process Synth 2019; 8: 742–755 Self-dual Leonard pairs Behzad Zeynizadeh, Farkhondeh Mohammad Aminzadeh and Hossein Mousavi* https://doi.org/10.1515/spma-2019-0001 Two different facileReceived and May efficient 8, 2018; accepted September approaches 22, 2018 for the synthesis ofAbstract: variousLet F denote N-arylacetamides a eld and let V denote a vector space over F with nite positive dimension. Consider a pair A, A∗ of diagonalizable F-linear maps on V, each of which acts on an eigenbasis for the other one in an via N-acetylation ofirreducible arylamines tridiagonal fashion. and Such a pair is called a Leonard pair. We consider the self-dual case in which there exists an automorphism of the endomorphism algebra of V that swaps A and A∗. Such an automorphism is unique, and called the duality A A∗. In the present paper we give a comprehensive description of this straightforward one-pot reductive acetylation↔ duality. In particular, we display an invertible F-linear map T on V such that the map X TXT− is the duality → A A∗. We express T as a polynomial in A and A∗. We describe how T acts on ags, decompositions, of nitroarenes promoted↔ by recyclable CuFe2O4 nanoparticles in waterand 24 bases for V. Keywords: Leonard pair, tridiagonal matrix, self-dual https://doi.org/10.1515/gps-2019-0044 Classication: 17B37,15A21their extensive applications in various area such as peptide Received December 17, 2018; accepted May 28, 2019. synthesis, agrochemicals, polymers, functional materials, dyes, fragrances and also existence in pharmaceuticals Abstract: Two simple, efficient, and environmentally (Figure 1) and natural products (Figure 2) [11-25]. -

Synthetic Efforts for Stereo Structure Determination of Cytotoxic Marine Natural Product Pericosines As Metabolites of Periconia Sp
Int. J. Mol. Sci. 2008, 9, 401-421 International Journal of Molecular Sciences ISSN 1422-0067 © 2008 by MDPI http://www.mdpi.org/ijms Review Synthetic Efforts for Stereo Structure Determination of Cytotoxic Marine Natural Product Pericosines as Metabolites of Periconia sp. from Sea Hare Yoshihide Usami,* Hayato Ichikawa and Masao Arimoto Osaka University of Pharmaceutical Sciences, 4-20-1 Nasahara, Takatsuki, Osaka 569-1094, Japan * Author to whom correspondence should be addressed; E-mail: [email protected] Received: 10 January 2008; in revised form: 18 March 2008 / Accepted: 19 March 2008 / Published: 24 March 2008 Abstract: Pericosines are unique C7 cyclohexenoid metabolites of Periconia byssoides OUPS-N133 fungus that was originally isolated from the sea hare, Aplysia kurodai. Pericosines show significant in vitro cytotoxicity against P388 lymphocytic leukemia cells. Pericosine A, in particular, shows the most potent activity and significant in vivo antitumor activity against P388 cells. Thus, pericosines are promising candidates for seed compounds of anticancer drugs. However, before the total syntheses of pericosines were accomplished, their stereo structures could not be determined by spectral analyses because they have multi- functionalized cyclohexenoid structures with torsional strain. In this review, synthetic efforts for pericosines in this decade are surveyed. Keywords: marine natural product, antitumor, pericosine, structure determination, total synthesis, carbasugar 1. Introduction Synthetic studies of carbasugars have been progressing on a worldwide scale [1-3]. Carbasugars are a class of carbocyclic analogues of monosaccharides in which oxygen atom in the ring is replaced with a carbon atom. Because of this, they are also called pseudo-sugars. Carbasugars exhibit gycosidase inhibitory, antitumor (including anticancer), antiviral, antifungal, antibacterial, and antimalarial activities. -

Chem 215 F11 Notes – Dr. Masato Koreeda - Page 1 of 14
Chem 215 F11 Notes – Dr. Masato Koreeda - Page 1 of 14. Date: September 30, 2011 Chapter 15: Carboxylic Acids and Their Derivatives – Acyl Transfer Reactions I. Introduction Examples: note: R could be "H" R Z R O H R O R' ester O carboxylic acid O O an acyl group bonded to R X R S acid halide* R' an electronegative atom (Z) thioester O X = halogen O R' R, R', R": alkyl, alkenyl, alkynyl, R O R' R N or aryl group R" amide O O O acid anhydride one of or both of R' and R" * acid halides could be "H" R F R Cl R Br R I O O O O acid fluoride acid chloride acid bromide acid iodide R Z sp2 hybridized; trigonal planar making it relatively "uncrowded" O The electronegative O atom polarizes the C=O group, making the C=O carbon "electrophilic." Resonance contribution by Z δ * R Z R Z R Z R Z C C C C O O O δ O hybrid structure The basicity and size of Z determine how much this resonance structure contributes to the hybrid. * The more basic Z is, the more it donates its electron pair, and the more resonance structure * contributes to the hybrid. similar basicity O R' Cl OH OR' NR'R" Trends in basicity: O weakest increasing basiciy strongest base base Check the pKa values of the conjugate acids of these bases. Chem 215 F11 Notes –Dr. Masato Koreeda - Page 2 of 14. Date: September 30, 2011 Relative stabilities of carboxylic acid derivatives against nucleophiles R Z As the basicity of Z increases, the stability of increases because of added resonance stabilization. -

Selective N-Acylation of Amino Alcohols Selektive N-Acylation Von Aminoalkoholen N-Acylation Selective D'amino-Alcools
Europaisches Patentamt (19) European Patent Office Office europeenpeen des brevets EP 0 633 875 B1 (12) EUROPEAN PATENT SPECIFICATION (45) Date of publication and mention (51) intci.6: C07C 231/02, C07C 309/00, of the grant of the patent: C07C 233/18, C07C 233/20, 02.01.1997 Bulletin 1997/01 C07C 235/08 (21) Application number: 93908917.3 (86) International application number: PCT/EP93/00849 (22) Date of filing: 02.04.1993 (87) International publication number: WO 93/20038 (14.10.1993 Gazette 1993/25) (54) SELECTIVE N-ACYLATION OF AMINO ALCOHOLS SELEKTIVE N-ACYLATION VON AMINOALKOHOLEN N-ACYLATION SELECTIVE D'AMINO-ALCOOLS (84) Designated Contracting States: (56) References cited: AT BE CH DE DK ES FR GB GR IE IT LI LU MC NL EP-A- 0 023 453 EP-A- 0 187 702 PT SE EP-A- 0 255 443 EP-A- 0 398 340 (30) Priority: 03.04.1992 EP 92200968 JOURNAL OF ORGANIC CHEMISTRY, vol. 56, no. 17, 16 August 1991, E ASTON US pages 5132 (43) Date of publication of application: - 8 M. BARTRA ET. AL. 'Cyclisation of 18.01.1995 Bulletin 1995/03 9-substituted decanoic acid derivatives to 9-decanolide and 9-decanelactam' (73) Proprietor: GIST-BROCADES N.V. CHEMICAL ABSTRACTS, vol. 86, no. 13, 28 NL-2600 MA Delft (NL) March 1977, Columbus, Ohio, US; abstract no. 891 25r, J.F.W. KEANA ET. AL. 'Stearoyl (72) Inventors: p-toluenesulfonate. A powerful acylating agent • SMEETS, Jan, Willem, Hubert for lipid synthesis.' page 484 ;column 1 ; NL-3732 GJ De Bilt (NL) CHEMISTRY AND PHYSICS OF LIPIDS vol. -

Nucleophilic Dearomatization of Activated Pyridines
Review Nucleophilic Dearomatization of Activated Pyridines Giulio Bertuzzi *, Luca Bernardi * and Mariafrancesca Fochi * Department of Industrial Chemistry “Toso Montanari” and INSTM RU Bologna, Alma Mater Studiorum-University of Bologna, Via Risorgimento 4, 40136 Bologna, Italy * Correspondence: [email protected] (G.B.); [email protected] (L.B.); [email protected] (M.F.); Tel.: +39-051-209-3626 (M.F.) Received: 16 November 2018; Accepted: 1 December 2018; Published: 6 December 2018 Abstract: Amongst nitrogen heterocycles of different ring sizes and oxidation statuses, dihydropyridines (DHP) occupy a prominent role due to their synthetic versatility and occurrence in medicinally relevant compounds. One of the most straightforward synthetic approaches to polysubstituted DHP derivatives is provided by nucleophilic dearomatization of readily assembled pyridines. In this article, we collect and summarize nucleophilic dearomatization reactions of - pyridines reported in the literature between 2010 and mid-2018, complementing and updating previous reviews published in the early 2010s dedicated to various aspects of pyridine chemistry. Since functionalization of the pyridine nitrogen, rendering a (transient) pyridinium ion, is usually required to render the pyridine nucleus sufficiently electrophilic to suffer the attack of a nucleophile, the material is organized according to the type of N-functionalization. A variety of nucleophilic species (organometallic reagents, enolates, heteroaromatics, umpoled aldehydes) can be productively engaged in pyridine dearomatization reactions, including catalytic asymmetric implementations, providing useful and efficient synthetic platforms to (enantioenriched) DHPs. Conversely, pyridine nitrogen functionalization can also lead to pyridinium ylides. These dipolar species can undergo a variety of dipolar cycloaddition reactions with electron-poor dipolarophiles, affording polycyclic frameworks and embedding a DHP moiety in their structures. -
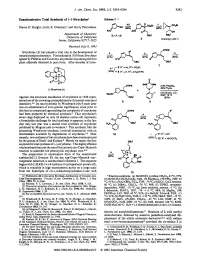
Enantioselective Total Synthesis of (-)-Strychnine1 Scheme1
J. Am. Chem. SOC.1993,115, 9293-9294 9293 Enantioselective Total Synthesis of (-)-Strychnine1 Scheme1 Steven D. Knight, Larry E. Overman,' and Garry Pairaudeau Department of Chemistry University of California Imine, California 9271 7-2025 Received July 8, 1993 OTlPS Strychnine (1) has played a vital role in the development of d h,i natural productschemistry. First isolated in 1818 fromstrychnos ignatii by Pelletier and Caventou, strychnine was among the first plant alkaloids obtained in pure form. After decades of inves- OBU' (-)-Strychnine(1) tigation, the structural elucidation of strychnine in 1946 repre- sented one of the crowning accomplishments of classical structural chemistry.394 Its total synthesis by Woodward only 8 years later was an achievement of even greater significance, since prior to this feat no compound approaching the complexity of strychnine ,oms had been prepared by chemical synthesis.* That strychnine's P' seven rings displayed on only 24 skeletal atoms still represents a formidable challenge for total synthesis is apparent in the fact that only last year was a second total synthesis of strychnine published by Magnus and co-workers.6 This synthesis, like the pioneering Woodward synthesis, involved intersection with an intermediate available by degradation of stry~hnine.~.~Most recently, two syntheses of (*)-strychnine have been communicated by the groups of Stork' and Kuehne.* Herein we report the first asymmetric total synthesis of (-)-strychnine. This highly efficient total synthesis features the use of the cationic aza-Cope-Mannich reaction to assemble the pentacyclic strychnan c~re.~J~ The preparation in enantiopure form of the unsaturated azabicyclo[3.2.