16.4 Electrophilic Aromatic Substitution Reactions of Benzene 751
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

De Novo Biosynthesis of Terminal Alkyne-Labeled Natural Products
De novo biosynthesis of terminal alkyne-labeled natural products Xuejun Zhu1,2, Joyce Liu2,3, Wenjun Zhang1,2,4* 1Department of Chemical and Biomolecular Engineering, 2Energy Biosciences Institute, 3Department of Bioengineering, University of California, Berkeley, CA 94720, USA. 4Physical Biosciences Division, Lawrence Berkeley National Laboratory, Berkeley, CA 94720, USA. *e-mail:[email protected] 1 Abstract: The terminal alkyne is a functionality widely used in organic synthesis, pharmaceutical science, material science, and bioorthogonal chemistry. This functionality is also found in acetylenic natural products, but the underlying biosynthetic pathways for its formation are not well understood. Here we report the characterization of the first carrier protein- dependent terminal alkyne biosynthetic machinery in microbes. We further demonstrate that this enzymatic machinery can be exploited for the in situ generation and incorporation of terminal alkynes into two natural product scaffolds in E. coli. These results highlight the prospect for tagging major classes of natural products, including polyketides and polyketide/non-ribosomal peptide hybrids, using biosynthetic pathway engineering. 2 Natural products are important small molecules widely used as drugs, pesticides, herbicides, and biological probes. Tagging natural products with a unique chemical handle enables the visualization, enrichment, quantification, and mode of action study of natural products through bioorthogonal chemistry1-4. One prevalent bioorthogonal reaction is -
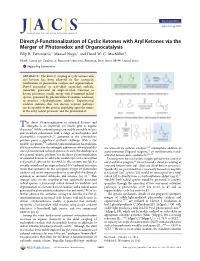
Direct Β‑Functionalization of Cyclic Ketones with Aryl Ketones Via the Merger of Photoredox and Organocatalysis Filip R
Communication pubs.acs.org/JACS Direct β‑Functionalization of Cyclic Ketones with Aryl Ketones via the Merger of Photoredox and Organocatalysis Filip R. Petronijevic,́† Manuel Nappi,† and David W. C. MacMillan* Merck Center for Catalysis at Princeton University, Princeton, New Jersey 08544, United States *S Supporting Information ABSTRACT: The direct β-coupling of cyclic ketones with aryl ketones has been achieved via the synergistic combination of photoredox catalysis and organocatalysis. Diaryl oxymethyl or aryl−alkyl oxymethyl radicals, transiently generated via single-electron reduction of ketone precursors, readily merge with β-enaminyl radical species, generated by photon-induced enamine oxidation, to produce γ-hydroxyketone adducts. Experimental evidence indicates that two discrete reaction pathways can be operable in this process depending upon the nature of the ketyl radical precursor and the photocatalyst. he direct β-functionalization of saturated ketones and T aldehydes is an important yet elusive goal in organic chemistry.1 While carbonyl groups are readily amenable to ipso- and α-carbon substitution with a range of nucleophiles and electrophiles respectively,2,3 activation at the β-methylene position poses a significant synthetic challenge. With a few notable exceptions,1,4 carbonyl β-functionalization has tradition- ally been restricted to the conjugate addition of soft nucleophiles are accessed via carbene catalysis,9,10 nucleophilic addition of into α,β-unsaturated carbonyl systems. As such, the development acetal-protected Grignard reagents,11 or stoichiometric metal- 5 − of a general catalytic platform for the direct β-functionalization activated homoenolate equivalents.12 16 of saturated ketones or aldehydes would represent a conceptual Drawing from the mechanistic insights gained in the course of and practical advance for the field. -

Phospholipid:Diacylglycerol Acyltransferase: an Enzyme That Catalyzes the Acyl-Coa-Independent Formation of Triacylglycerol in Yeast and Plants
Phospholipid:diacylglycerol acyltransferase: An enzyme that catalyzes the acyl-CoA-independent formation of triacylglycerol in yeast and plants Anders Dahlqvist*†‡, Ulf Ståhl†§, Marit Lenman*, Antoni Banas*, Michael Lee*, Line Sandager¶, Hans Ronne§, and Sten Stymne¶ *Scandinavian Biotechnology Research (ScanBi) AB, Herman Ehles Va¨g 2 S-26831 Svaloˆv, Sweden; ¶Department of Plant Breeding Research, Swedish University of Agricultural Sciences, Herman Ehles va¨g 2–4, S-268 31 Svalo¨v, Sweden; and §Department of Plant Biology, Uppsala Genetic Center, Swedish University of Agricultural Sciences, Box 7080, S-750 07 Uppsala, Sweden Edited by Christopher R. Somerville, Carnegie Institution of Washington, Stanford, CA, and approved March 31, 2000 (received for review February 15, 2000) Triacylglycerol (TAG) is known to be synthesized in a reaction that acid) and epoxidated fatty acid (vernolic acid) in TAG in castor uses acyl-CoA as acyl donor and diacylglycerol (DAG) as acceptor, bean (Ricinus communis) and the hawk’s-beard Crepis palaestina, and which is catalyzed by the enzyme acyl-CoA:diacylglycerol respectively. Furthermore, a similar enzyme is shown to be acyltransferase. We have found that some plants and yeast also present in the yeast Saccharomyces cerevisiae, and the gene have an acyl-CoA-independent mechanism for TAG synthesis, encoding this enzyme, YNR008w, is identified. which uses phospholipids as acyl donors and DAG as acceptor. This reaction is catalyzed by an enzyme that we call phospholipid:dia- Materials and Methods cylglycerol acyltransferase, or PDAT. PDAT was characterized in Yeast Strains and Plasmids. The wild-type yeast strains used were microsomal preparations from three different oil seeds: sunflower, either FY1679 (MAT␣ his3-⌬200 leu2-⌬1 trp1-⌬6 ura3-52) (9) or castor bean, and Crepis palaestina. -

Common Name: P-TOLUENE SULFONIC ACID HAZARD
Common Name: p-TOLUENE SULFONIC ACID CAS Number: 104-15-4 DOT Number: UN 2583 (solid, with more than 5% free Sulfuric Acid) UN 2585 (solid, with less than 5% free RTK Substance number: 1870 Sulfuric Acid) Date: October 1996 Revision: May 2003 ------------------------------------------------------------------------- ------------------------------------------------------------------------- HAZARD SUMMARY WORKPLACE EXPOSURE LIMITS * p-Toluene Sulfonic Acid can affect you when breathed in. No occupational exposure limits have been established for * p-Toluene Sulfonic Acid is a CORROSIVE CHEMICAL p-Toluene Sulfonic Acid. This does not mean that this and contact can cause severe skin and eye irritation and substance is not harmful. Safe work practices should always burns. be followed. * Exposure to p-Toluene Sulfonic Acid can irritate the nose, throat and lungs causing burning, dryness and WAYS OF REDUCING EXPOSURE coughing. * Where possible, enclose operations and use local exhaust ventilation at the site of chemical release. If local exhaust IDENTIFICATION ventilation or enclosure is not used, respirators should be p-Toluene Sulfonic Acid is a colorless, clear, or dark-colored worn. liquid or a colorless, crystalline (sand-like) material. It is used * Wear protective work clothing. to make dyes, drugs and other chemicals. * Wash thoroughly immediately after exposure to p- Toluene Sulfonic Acid and at the end of the workshift. REASON FOR CITATION * Post hazard and warning information in the work area. In * p-Toluene Sulfonic Acid is on the Hazardous Substance addition, as part of an ongoing education and training List because it is cited by DOT and NFPA. effort, communicate all information on the health and * This chemical is on the Special Health Hazard Substance safety hazards of p-Toluene Sulfonic Acid to potentially List because it is CORROSIVE. -

Products from Reactions of Carbon Nucleophiles and Carbon
14C synthesis strategies, Chem 315/316 / Beauchamp 1 Products from reactions of carbon nucleophiles and carbon electrophiles used in the 14C Game and our course: Carbon and hydrogen nucleophiles Ph R Ph Al H N R Li R (MgBr) P Li AlH R Cu Li Na CN RCC Na Ph 4 Li Carbon 2 H C R organolithium organolithium cyanide acetylides 2 ylid Na BH4 diisobutylaluminium lithium diisopropy- electrophiles cuprates (LAH) o reagents reagents Wittig reagents hydride (DIBAH) amide (LDA), -78 C H C Br 3 not useful not useful 2 RX coupling nitrilesalkynes not useful alkyls alkyls not useful methyl RX reaction R Br not useful not useful 2 RX coupling nitriles alkynes not useful alkyls alkyls not useful primary RX reaction R 2 RX coupling nitriles alkyls not useful E2 not useful alkyls not useful not useful reaction R Br secondary RX O 1o ROH 1o ROH 1o ROH 1o ROH not useful 1o ROH o not useful not useful 1o ROH 1 ROH ethylene oxide nitriles alkynes O o o o o o o 2 ROH 2 ROH 2 ROH 2 ROH 2o ROH 2 ROH 2 ROH not useful not useful E2, make nitriles alkynes allylic alcohols propylene oxide O o 3 ROH 3o ROH o 3o ROH o not useful o 3o ROH not useful E2, make 3 ROH 3 ROH 3 ROH allylic alcohols nitriles alkynes isobutylene oxide O o o specific methanol methanol C 1 ROH 1 ROH not used cyanohydrin 1o ROH not useful not useful in our course alkenes H H alkynes methanal O specific o enolate o o cyanohydrin o 1 ROH o C 2 ROH 2 ROH not useful 2 ROH alkenes 1 ROH not useful chemistry R H alkynes simple aldehydes O cyanohydrin o unless specific o enolate C 3 ROH 3o ROH o 2o ROH -

Organic Chemistry
Wisebridge Learning Systems Organic Chemistry Reaction Mechanisms Pocket-Book WLS www.wisebridgelearning.com © 2006 J S Wetzel LEARNING STRATEGIES CONTENTS ● The key to building intuition is to develop the habit ALKANES of asking how each particular mechanism reflects Thermal Cracking - Pyrolysis . 1 general principles. Look for the concepts behind Combustion . 1 the chemistry to make organic chemistry more co- Free Radical Halogenation. 2 herent and rewarding. ALKENES Electrophilic Addition of HX to Alkenes . 3 ● Acid Catalyzed Hydration of Alkenes . 4 Exothermic reactions tend to follow pathways Electrophilic Addition of Halogens to Alkenes . 5 where like charges can separate or where un- Halohydrin Formation . 6 like charges can come together. When reading Free Radical Addition of HX to Alkenes . 7 organic chemistry mechanisms, keep the elec- Catalytic Hydrogenation of Alkenes. 8 tronegativities of the elements and their valence Oxidation of Alkenes to Vicinal Diols. 9 electron configurations always in your mind. Try Oxidative Cleavage of Alkenes . 10 to nterpret electron movement in terms of energy Ozonolysis of Alkenes . 10 Allylic Halogenation . 11 to make the reactions easier to understand and Oxymercuration-Demercuration . 13 remember. Hydroboration of Alkenes . 14 ALKYNES ● For MCAT preparation, pay special attention to Electrophilic Addition of HX to Alkynes . 15 Hydration of Alkynes. 15 reactions where the product hinges on regio- Free Radical Addition of HX to Alkynes . 16 and stereo-selectivity and reactions involving Electrophilic Halogenation of Alkynes. 16 resonant intermediates, which are special favor- Hydroboration of Alkynes . 17 ites of the test-writers. Catalytic Hydrogenation of Alkynes. 17 Reduction of Alkynes with Alkali Metal/Ammonia . 18 Formation and Use of Acetylide Anion Nucleophiles . -

Aldehydes and Ketones
12 Aldehydes and Ketones Ethanol from alcoholic beverages is first metabolized to acetaldehyde before being broken down further in the body. The reactivity of the carbonyl group of acetaldehyde allows it to bind to proteins in the body, the products of which lead to tissue damage and organ disease. Inset: A model of acetaldehyde. (Novastock/ Stock Connection/Glow Images) KEY QUESTIONS 12.1 What Are Aldehydes and Ketones? 12.8 What Is Keto–Enol Tautomerism? 12.2 How Are Aldehydes and Ketones Named? 12.9 How Are Aldehydes and Ketones Oxidized? 12.3 What Are the Physical Properties of Aldehydes 12.10 How Are Aldehydes and Ketones Reduced? and Ketones? 12.4 What Is the Most Common Reaction Theme of HOW TO Aldehydes and Ketones? 12.1 How to Predict the Product of a Grignard Reaction 12.5 What Are Grignard Reagents, and How Do They 12.2 How to Determine the Reactants Used to React with Aldehydes and Ketones? Synthesize a Hemiacetal or Acetal 12.6 What Are Hemiacetals and Acetals? 12.7 How Do Aldehydes and Ketones React with CHEMICAL CONNECTIONS Ammonia and Amines? 12A A Green Synthesis of Adipic Acid IN THIS AND several of the following chapters, we study the physical and chemical properties of compounds containing the carbonyl group, C O. Because this group is the functional group of aldehydes, ketones, and carboxylic acids and their derivatives, it is one of the most important functional groups in organic chemistry and in the chemistry of biological systems. The chemical properties of the carbonyl group are straightforward, and an understanding of its characteristic reaction themes leads very quickly to an understanding of a wide variety of organic reactions. -

Chapter 7, Haloalkanes, Properties and Substitution Reactions of Haloalkanes Table of Contents 1
Chapter 7, Haloalkanes, Properties and Substitution Reactions of Haloalkanes Table of Contents 1. Alkyl Halides (Haloalkane) 2. Nucleophilic Substitution Reactions (SNX, X=1 or 2) 3. Nucleophiles (Acid-Base Chemistry, pka) 4. Leaving Groups (Acid-Base Chemistry, pka) 5. Kinetics of a Nucleophilic Substitution Reaction: An SN2 Reaction 6. A Mechanism for the SN2 Reaction 7. The Stereochemistry of SN2 Reactions 8. A Mechanism for the SN1 Reaction 9. Carbocations, SN1, E1 10. Stereochemistry of SN1 Reactions 11. Factors Affecting the Rates of SN1 and SN2 Reactions 12. --Eliminations, E1 and E2 13. E2 and E1 mechanisms and product predictions In this chapter we will consider: What groups can be replaced (i.e., substituted) or eliminated The various mechanisms by which such processes occur The conditions that can promote such reactions Alkyl Halides (Haloalkane) An alkyl halide has a halogen atom bonded to an sp3-hybridized (tetrahedral) carbon atom The carbon–chlorine and carbon– bromine bonds are polarized because the halogen is more electronegative than carbon The carbon-iodine bond do not have a per- manent dipole, the bond is easily polarizable Iodine is a good leaving group due to its polarizability, i.e. its ability to stabilize a charge due to its large atomic size Generally, a carbon-halogen bond is polar with a partial positive () charge on the carbon and partial negative () charge on the halogen C X X = Cl, Br, I Different Types of Organic Halides Alkyl halides (haloalkanes) sp3-hybridized Attached to Attached to Attached -

Chemical Kinetics HW1 (Kahn, 2010)
Chemical Kinetics HW1 (Kahn, 2010) Question 1. (6 pts) A reaction with stoichiometry A = P + 2Q was studied by monitoring the concentration of the reactant A as a function of time for eighteen minutes. The concentration determination method had a maximum error of 6 M. The following concentration profile was observed: Time (min) Conc (mM) 1 0.9850 2 0.8571 3 0.7482 4 0.6549 5 0.5885 6 0.5183 7 0.4667 8 0.4281 9 0.3864 10 0.3557 11 0.3259 12 0.3037 13 0.2706 14 0.2486 15 0.2355 16 0.2188 17 0.2111 18 0.1930 Determine the reaction order and calculate the rate constant for decomposition of A. What can be said about the mechanism or molecularity of this reaction? Question 2. (4 pts) Solve problem 2 on pg 31 in your textbook (House) using both linear and non-linear regression. Provide standard errors for the rate constant and half-life based on linear and non-linear fits. Below is the data set for your convenience: dataA = {{0, 0.5}, {10, 0.443}, {20,0.395}, {30,0.348}, {40,0.310}, {50,0.274}, {60,0.24}, {70,0.212}, {80,0.190}, {90,0.171}, {100,0.164}} Question 3. (3 pts) Solve problem 3 on pg 32 in your textbook (House). Question 4. (7 pts) The authors of the paper “Microsecond Folding of the Cold Shock Protein Measured by a Pressure-Jump Technique” suggest that the activated state of folding of CspB follows Hammond- type behavior. -

Structural Determination of Subsidiary Colors in Commercial Food Blue No
February 1998 7 Original Structural Determination of Subsidiary Colors in Commercial Food Blue No. 1 (Brilliant Blue FCF) Product (Received September 1, 1997) Hirosh1 MATSUFUJI*1, Takashi KUSAKA*1, Masatoshi TSUKUDA*1, Makoto CHINO*1, Yoshiaki KATO*2, Mikio NAKAMURA*2, Yukihiro GODA*3, Masatake TOYODA*3 and Mitsuharu TAKEDA*1 (*1College of Bioresource Sciences, Nihon University: 3-34-1, Shimouma, Setagaya-ku, Tokyo 145-0002, Japan; *2San-Ei Gen F. F. I., Inc.: 1-1-11, Sanwa-cho, Toyonaka, Osaka 561-0828, Japan; *3National Institute of Health Sciences (NIHS): 1-18-1, Kamiyoga, Setagaya-ku, Tokyo 158-8501, Japan) HPLC analysis revealed that five subsidiary colors were present in a commercial Food Blue No. l (Brilliant Blue FCF) product. Among them, major subsidiary colors C, D, and E were isolated. On the bases of spectroscopic analyses, their structures were identified as the disodium salt of 2-[[4-[N-ethyl-N-(3-sulfophenylmethyl)amino]phenyl][4-[N-ethyl-N-(4-sulfo- phenylmethyl)amino]phenyl]methylio]benzenesulfonic acid, the disodium salt of 2-[[4- [N-ethyl-N-(2-sulfophenylmethyl) amino]phenyl][4-[N-ethyl-N-(3-sulfophenylmethyl) amino]- phenyl]methylio]benzenesulfonic acid, and the sodium salt of 2-[[4-(N-ethylamino)phenyl][4- [N-ethyl-N-(3-sulfophenylmethyl)amino]phenyl]methylio]benzenesulfonic acid, respectively. Key words: Food Blue No. 1; Brilliant Blue FCF; FD & C Blue No. 1; subsidiary color; HPLC; coal-tar dye ally, it is the disodium salt of 2-[bis[4-[N-ethyl- Introduction N-(3-sulfophenylmethyl) amino] phenyl] meth- Twelve coal-tar dyes are presently permitted ylio]benzensulfonic acid [OSBA-(m-EBASA)(m- as food colors in Japan. -

Course Material 2.Pdf
Reactive Intermediates Source: https://www.askiitians.com/iit-jee-chemistry/organic-chemistry/iupac- and-goc/reaction-intermediates/ Table of Content • Carbocations • Carbanions • Free Radicals • Carbenes • Arenium Ions • Benzynes Synthetic intermediate are stable products which are prepared, isolated and purified and subsequently used as starting materials in a synthetic sequence. Reactive intermediate, on the other hand, are short lived and their importance lies in the assignment of reaction mechanisms on the pathway from the starting substrate to stable products. These reactive intermediates are not isolated, but are detected by spectroscopic methods, or trapped chemically or their presence is confirmed by indirect evidence. • Carbocations Carbocations are the key intermediates in several reactions and particularly in nucleophilic substitution reactions. Structure of Carbocations : Generally, in the carbocations the positively charged carbon atom is bonded to three other atoms and has no nonbonding electrons. It is sp 2 hybridized with a planar structure and bond angles of about 120°. There is a + vacant unhybridized p orbital which in the case of CH 3 lies perpendicular to the plane of C—H bonds. Stability of Carbocations: There is an increase in carbocation stability with additional alkyl substitution. Thus one finds that addition of HX to three typical olefins decreases in the order (CH 3)2C=CH 2>CH 3—CH = CH 2 > CH 2 = CH 2. This is due to the relative stabilities of the carbocations formed in the rate determining step which in turn follows from the fact that the stability is increased by the electron releasing methyl group (+I), three such groups being more effective than two, and two more effective than one. -

Reactions of Benzene & Its Derivatives
Organic Lecture Series ReactionsReactions ofof BenzeneBenzene && ItsIts DerivativesDerivatives Chapter 22 1 Organic Lecture Series Reactions of Benzene The most characteristic reaction of aromatic compounds is substitution at a ring carbon: Halogenation: FeCl3 H + Cl2 Cl + HCl Chlorobenzene Nitration: H2 SO4 HNO+ HNO3 2 + H2 O Nitrobenzene 2 Organic Lecture Series Reactions of Benzene Sulfonation: H 2 SO4 HSO+ SO3 3 H Benzenesulfonic acid Alkylation: AlX3 H + RX R + HX An alkylbenzene Acylation: O O AlX H + RCX 3 CR + HX An acylbenzene 3 Organic Lecture Series Carbon-Carbon Bond Formations: R RCl AlCl3 Arenes Alkylbenzenes 4 Organic Lecture Series Electrophilic Aromatic Substitution • Electrophilic aromatic substitution: a reaction in which a hydrogen atom of an aromatic ring is replaced by an electrophile H E + + + E + H • In this section: – several common types of electrophiles – how each is generated – the mechanism by which each replaces hydrogen 5 Organic Lecture Series EAS: General Mechanism • A general mechanism slow, rate + determining H Step 1: H + E+ E El e ctro - Resonance-stabilized phile cation intermediate + H fast Step 2: E + H+ E • Key question: What is the electrophile and how is it generated? 6 Organic Lecture Series + + 7 Organic Lecture Series Chlorination Step 1: formation of a chloronium ion Cl Cl + + - - Cl Cl+ Fe Cl Cl Cl Fe Cl Cl Fe Cl4 Cl Cl Chlorine Ferric chloride A molecular complex An ion pair (a Lewis (a Lewis with a positive charge containing a base) acid) on ch lorine ch loronium ion Step 2: attack of