Ultrapotent Vinblastines in Which Added Molecular Complexity Further
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Modifications on the Basic Skeletons of Vinblastine and Vincristine
Molecules 2012, 17, 5893-5914; doi:10.3390/molecules17055893 OPEN ACCESS molecules ISSN 1420-3049 www.mdpi.com/journal/molecules Review Modifications on the Basic Skeletons of Vinblastine and Vincristine Péter Keglevich, László Hazai, György Kalaus and Csaba Szántay * Department of Organic Chemistry and Technology, University of Technology and Economics, H-1111 Budapest, Szt. Gellért tér 4, Hungary * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel: +36-1-463-1195; Fax: +36-1-463-3297. Received: 30 March 2012; in revised form: 9 May 2012 / Accepted: 10 May 2012 / Published: 18 May 2012 Abstract: The synthetic investigation of biologically active natural compounds serves two main purposes: (i) the total synthesis of alkaloids and their analogues; (ii) modification of the structures for producing more selective, more effective, or less toxic derivatives. In the chemistry of dimeric Vinca alkaloids enormous efforts have been directed towards synthesizing new derivatives of the antitumor agents vinblastine and vincristine so as to obtain novel compounds with improved therapeutic properties. Keywords: antitumor therapy; vinblastine; vincristine; derivatives 1. Introduction Vinblastine (1) and vincristine (2) are dimeric alkaloids (Figure 1) isolated from the Madagaskar periwinkle plant (Catharantus roseus), exhibit significant cytotoxic activity and are used in the antitumor therapy as antineoplastic agents. In the course of cell proliferation they act as inhibitors during the metaphase of the cell cycle and by binding to the microtubules inhibit the development of the mitotic spindle. In tumor cells these agents inhibit the DNA repair and the RNA synthesis mechanisms, blocking the DNA-dependent RNA polymerase. Molecules 2012, 17 5894 Figure 1. -
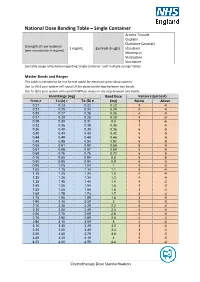
National Dose Banding Table – Single Container
National Dose Banding Table – Single Container Arsenic Trioxide Cisplatin Cladribine (Leustat) Strength of raw material 1 mg/mL Example drug(s) Idarubicin (after reconstitution if required) Mitomycin Vinblastine Vincristine See table usage notes below regarding ‘single container’ and ‘multiple syringe’ tables. Master Bands and Ranges This table is intended to be in a format useful for electronic prescribing systems. Use To (A) if your system will round UP for doses on the step between two bands. Use To (B) if your system will round DOWN for doses on the step between two bands. Band Range (mg) Band Dose Variance (percent) From ≥ To (A) < To (B) ≤ (mg) Below Above 0.21 0.23 0.22 0.22 5 -4 0.23 0.25 0.24 0.24 4 -4 0.25 0.27 0.26 0.26 4 -4 0.27 0.29 0.28 0.28 4 -3 0.29 0.32 0.31 0.3 3 -6 0.32 0.36 0.35 0.34 7 -5 0.36 0.40 0.39 0.38 6 -5 0.40 0.44 0.43 0.42 5 -5 0.44 0.49 0.48 0.46 5 -6 0.49 0.55 0.54 0.52 6 -5 0.55 0.61 0.60 0.58 6 -5 0.61 0.68 0.67 0.64 5 -6 0.68 0.76 0.75 0.72 6 -5 0.76 0.85 0.84 0.8 5 -6 0.85 0.95 0.94 0.9 6 -5 0.95 1.05 1.04 1 5 -5 1.05 1.15 1.14 1.1 5 -4 1.15 1.25 1.24 1.2 4 -4 1.25 1.35 1.34 1.3 4 -4 1.35 1.45 1.44 1.4 4 -3 1.45 1.55 1.54 1.5 4 -3 1.55 1.65 1.64 1.6 3 -3 1.65 1.75 1.74 1.7 3 -3 1.75 1.90 1.89 1.8 3 -5 1.90 2.10 2.09 2 5 -5 2.10 2.30 2.29 2.2 5 -4 2.30 2.50 2.49 2.4 4 -4 2.50 2.70 2.69 2.6 4 -4 2.70 2.90 2.89 2.8 4 -3 2.90 3.10 3.09 3 4 -3 3.10 3.30 3.29 3.2 3 -3 3.30 3.50 3.49 3.4 3 -3 3.50 3.80 3.79 3.6 3 -5 3.80 4.20 4.19 4 5 -5 4.20 4.60 4.59 4.4 5 -4 Chemotherapy Dose Standardisation Band Range (mg) -

Hodgkin Lymphoma Treatment Regimens
HODGKIN LYMPHOMA TREATMENT REGIMENS (Part 1 of 5) Clinical Trials: The National Comprehensive Cancer Network recommends cancer patient participation in clinical trials as the gold standard for treatment. Cancer therapy selection, dosing, administration, and the management of related adverse events can be a complex process that should be handled by an experienced health care team. Clinicians must choose and verify treatment options based on the individual patient; drug dose modifications and supportive care interventions should be administered accordingly. The cancer treatment regimens below may include both U.S. Food and Drug Administration-approved and unapproved indications/regimens. These regimens are provided only to supplement the latest treatment strategies. These Guidelines are a work in progress that may be refined as often as new significant data become available. The NCCN Guidelines® are a consensus statement of its authors regarding their views of currently accepted approaches to treatment. Any clinician seeking to apply or consult any NCCN Guidelines® is expected to use independent medical judgment in the context of individual clinical circumstances to determine any patient’s care or treatment. The NCCN makes no warranties of any kind whatsoever regarding their content, use, or application and disclaims any responsibility for their application or use in any way. Classical Hodgkin Lymphoma1 Note: All recommendations are Category 2A unless otherwise indicated. Primary Treatment Stage IA, IIA Favorable (No Bulky Disease, <3 Sites of Disease, ESR <50, and No E-lesions) REGIMEN DOSING Doxorubicin + Bleomycin + Days 1 and 15: Doxorubicin 25mg/m2 IV push + bleomycin 10units/m2 IV push + Vinblastine + Dacarbazine vinblastine 6mg/m2 IV over 5–10 minutes + dacarbazine 375mg/m2 IV over (ABVD) (Category 1)2-5 60 minutes. -

BC Cancer Benefit Drug List September 2021
Page 1 of 65 BC Cancer Benefit Drug List September 2021 DEFINITIONS Class I Reimbursed for active cancer or approved treatment or approved indication only. Reimbursed for approved indications only. Completion of the BC Cancer Compassionate Access Program Application (formerly Undesignated Indication Form) is necessary to Restricted Funding (R) provide the appropriate clinical information for each patient. NOTES 1. BC Cancer will reimburse, to the Communities Oncology Network hospital pharmacy, the actual acquisition cost of a Benefit Drug, up to the maximum price as determined by BC Cancer, based on the current brand and contract price. Please contact the OSCAR Hotline at 1-888-355-0355 if more information is required. 2. Not Otherwise Specified (NOS) code only applicable to Class I drugs where indicated. 3. Intrahepatic use of chemotherapy drugs is not reimbursable unless specified. 4. For queries regarding other indications not specified, please contact the BC Cancer Compassionate Access Program Office at 604.877.6000 x 6277 or [email protected] DOSAGE TUMOUR PROTOCOL DRUG APPROVED INDICATIONS CLASS NOTES FORM SITE CODES Therapy for Metastatic Castration-Sensitive Prostate Cancer using abiraterone tablet Genitourinary UGUMCSPABI* R Abiraterone and Prednisone Palliative Therapy for Metastatic Castration Resistant Prostate Cancer abiraterone tablet Genitourinary UGUPABI R Using Abiraterone and prednisone acitretin capsule Lymphoma reversal of early dysplastic and neoplastic stem changes LYNOS I first-line treatment of epidermal -

Transitional Cell Carcinoma: Options Beyond Nsaids Julie Marie Gillem, DVM, DACVIM (Oncology) Overview
Transitional Cell Carcinoma: Options Beyond NSAIDs Julie Marie Gillem, DVM, DACVIM (Oncology) Overview ✦ Background ✦ Surgical Options ✦ Pathology ✦ Medical Options ✦ Location and staging ✦ Radiation Therapy ✦ Behavior Options ✦ Etiology and risk factors ✦ Palliative care ✦ Work up and diagnosis ✦ What about cats? Objectives ✦ How do we determine when NSAIDs fail? ✦ When should we intervene with surgery, chemotherapy, radiation therapy, and additional palliative care? Pathology ✦ ~2% of canine cancer ✦ Invasive transitional cell carcinoma (TCC) most common ✦ Others: SCC, adenocarcinoma, undifferentiated carcinoma, rhabdomyosarcoma, fibroma, and other mesenchymal tumors Location and Staging ✦ TCC in dogs most often found in the trigone of the bladder ✦ Series of 102 dogs at PUVTH ✦ Urethra and bladder in 56% ✦ Prostate involvement in 29% male dogs ✦ Lymph node mets in 16% at diagnosis ✦ Distant mets in 14% at diagnosis ✦ Distant mets in 50% at death Location ✦ TCC in dogs most often is found in the trigone region of the bladder. ✦ In a series of dogs with TCC examined at the PUVTH, the tumor involved the urethra as well as the bladder in 57 of 102 dogs (56%), and it involved the prostate in 11 of 38 (29%) male dogs. WHO Staging ✦ 78% T2 tumors ✦ 20% T3 tumors Biological Behavior ✦ At diagnosis: ✦ Regional lymph node metastasis in 12-46 % (Norris et al 1992, Knapp et al 2000, Blackburn et al 2013) ✦ Distant metastasis in 16- 23% (Norris et al 1992, Blackburn et al 2013) ✦ Distant metastasis in 50% at death (Norris et al 1992, Knapp et al -

Eliminating Vincristine Administration Events
Issue 37 October 2017 Eliminating vincristine administration events Issue: Despite the usually deadly consequences of accidentally administering the chemotherapy drug vincristine intrathecally, adverse events still occur, typically because some organizations still administer vincristine via syringe. The good news is that these events are happening less in the United States, mostly because of the efforts of leading national organizations to promote an effective prevention strategy that assures a mechanical barrier to intrathecal administration (into the subarachnoid space). The strategy involves diluting intravenous vincristine or other vinca alkaloids in a minibag that contains a volume that is too large for intrathecal administration (e.g., 25 mL for pediatric patients and 50 mL for adults), making it mechanically difficult to accidentally administer intrathecally.1 Vinca alkaloids (vinblastine, vinorelbine, vincristine, and vincristine liposomal) are chemotherapy drugs that are intended to be administered intravenously. If given intrathecally, vincristine is nearly always fatal and associated with an irreversible, painful ascending paralysis.2 When vinca alkaloids are injected intrathecally, destruction of the central nervous system occurs, radiating out from the injection site. The few survivors of this adverse event experienced devastating neurological damage.1 Part of the problem stems from ordering intravenous vincristine in conjunction with medications that are administered intrathecally via a syringe, such as methotrexate, cytarabine and hydrocortisone. In some adverse events, vincristine was mistakenly injected into the cerebrospinal fluid (CSF) of patients when the intent was to inject another intrathecal chemotherapy agent, such as methotrexate or cytarabine.3 Intravenous vincristine and other vinca alkaloids are dispensed from the pharmacy with explicit warning labels about their lethality if given intrathecally. -

Vincristine Archive File
Vincristine archive file Vincristine is a vinakaloid, which was first discovered 1958 in the tropical plant Catharanthus roseus, native in Madagascar. Its ability to inhibit the metaphase of the mitosis by suppressing the polymerization of the microtubule makes it very important as a chemotherapeutic agent, especially against non-Hodgkin lymphomas and Wilms' tumor. History: Vinblastine and Vincristine are bisindole alkaloids and both widely known for their use as antitumor drugs. In former times they were isolated in trace quantities from the leaves of Catharanthus roseus. Because of their importance in the medical field numerous researches were examining the structure, use and synthesis of Vinblastine, Vincristine and their derivates. Their biological effect to inhibit the microtubule formation and mitosis was and still is a very important part of medical cancer therapy. They were both among the first natural products whose structures were identified by X-ray crystallography and among the first for which X-ray analysis of a heavy atom derivative was used to establish their absolute configuration. ( 2) Timeline: 1958 Vinblastine was first discovered as an unexpected myelosuppressive agent by Noble, R. L., C. T. Beer, and J. H. Cutts during the search for an antidiabetic agent in Catharanthus roseus (myelosupression decreased activity of the bone marrow) 1959 independendently researchers from Eli Lilly (Johnson, I. S., J. G. Armstrong, M. Gorman, and J. P. Burnett, Jr.)discovered that the extracts of C. roseus Possesses activity against -

Differential Activity of Vincristine and Vinblastine Against Cultured Cells1 ;
[CANCER RESEARCH 44, 3307-3312, August 1984] Differential Activity of Vincristine and Vinblastine against Cultured Cells1 ; Peter J. Ferguson,2 J. Robert Phillips, Milada Seiner, and Carol E. Cass3 Cancer Research Group (McEachern Laboratory) [P. J. F., J. Ft. P., M. S., C. E. C.] and Department of Biochemistry [P. J. F., C. E. C.¡,University of Alberta, Edmonton, Alberta, Canada T6G 2H7 ABSTRACT determine if differential activity is related to differences in uptake4 or release of drug. Included were cells from these established Vincristine and vinblastine exhibit differential activity against lines: mouse neuroblastoma; mouse leukemia L1210; mouse tumors and normal tissues. In this work, a number of cultured lymphoma S49; HeLa; and human promyelocytic leukemia HL- cell lines were assayed for their sensitivity to the antiproliferative 60. Drug sensitivity in each cell line was determined by assaying and cytotoxic effects of the two drugs following short-term (4 hr) inhibition of proliferation during a continuous exposure to either or during continuous exposures. Differential activity was not drug or by assaying colony formation following a 24-hr exposure. seen when cells were subjected to continuous exposures. The Because of rapid loss of Vinca alkaloids from human serum concentrations of Vincristine and vinblastine, respectively, that following an i.v. bolus injection (11, 12,14, 17), the sensitivity of inhibited growth rates by 50% were: mouse leukemia L1210 cells to short-term (1- and 4-hr) exposures was also determined cells, 4.4 and 4.0 nw; mouse lymphoma S49 cells, 5 and 3.5 nM; for both drugs. Although there were differences in sensitivity to mouse neuroblastoma cells, 33 and 15 nw; HeLa cells, 1.4 and vincristine and vinblastine between cell lines, there was little or 2.6 nw; and human leukemia HL-60 cells, 4.1 and 5.3 nM. -

VINBLASTINE-VINCRISTINE (Chlvpp-EVA
Chemotherapy Protocol LYMPHOMA CHLORAMBUCIL-DOXORUBICIN-ETOPOSIDE-PREDNISOLONE-PROCARBAZINE- VINBLASTINE-VINCRISTINE (ChlVPP-EVA) Regimen • Lymphoma – ChlVPP-EVA-Chlorambucil-Doxorubicin-Etoposide-Prednisolone- Procarbazine-Vinblastine-Vincristine Indication • Hodgkin’s Lymphoma Toxicity Drug Adverse Effect Chlorambucil Gastro-intestinal disturbance Doxorubicin Cardiomyopathy, alopecia, urinary discolouration (red) Etoposide Hypotension on rapid infusion, hyperbilirubinaemia Weight gain, gastro-intestinal disturbances, hyperglycaemia, Prednisolone CNS disturbances, cushingoid changes, glucose intolerance Procarbazine Insomnia, ataxia, hallucinations, headache Vinblastine Peripheral neuropathy, constipation, jaw pain, ileus Vincristine Peripheral neuropathy, constipation, jaw pain, ileus The adverse effects listed are not exhaustive. Please refer to the relevant Summary of Product Characteristics for full details. Patients diagnosed with Hodgkin’s Lymphoma carry a lifelong risk of transfusion-associated graft versus host disease (TA-GVHD). Where blood products are required these patients must receive only irradiated blood products for life. Local blood transfusion departments must be notified as soon as a diagnosis is made and the patient must be issued with an alert card to carry with them at all times. Monitoring Drugs • FBC, LFTs and U&Es prior to day one and eight of treatment Version 1 (May 2018) Page 1 of 10 Lymphoma- ChlVPP-EVA-Chlorambucil-Doxorubicin-Etoposide-Prednisolone-Procarbazine-Vinblastine-Vincristine Dose Modifications The dose modifications listed are for haematological, liver and renal function and limited drug specific toxicities. Dose adjustments may be necessary for other toxicities as well. In principle all dose reductions due to adverse drug reactions should not be re-escalated in subsequent cycles without consultant approval. It is also a general rule for chemotherapy that if a third dose reduction is necessary treatment should be stopped. -

Combination Chemotherapy with Estramustine Phosphate, Ifosfamide and Cisplatin for Hormone-Refractory Prostate Cancer
Acta Med. Okayama, 2006 Vol. 60, No. 1, pp. 43ン49 CopyrightⒸ 2006 by Okayama University Medical School. Original Article http ://www.lib.okayama-u.ac.jp/www/acta/ Combination Chemotherapy with Estramustine Phosphate, Ifosfamide and Cisplatin for Hormone-refractory Prostate Cancer Haruki Kakua, Takashi Saikaa*, Tomoyasu Tsushimab, Atsushi Nagaia, Teruhiko Yokoyamaa, Fernando Abarzuaa, Shin Ebaraa, Daisuke Manabea, Yasutomo Nasua, and Hiromi Kumona aDepartment of Urology, Okayama University Graduate School of Medicine, Dentistry and Pharmaceutical Sciences, Okayama 700ン8558, Japan, and bDepartment of Urology, Medival center of Okayama, Okayama 701-1192, Japan We evaluated the effi ciency and toxicity of estramustine phosphate (ECT), ifosfamide (IFM) and cis- platin (CDDP) combination chemotherapy in twenty-one patients with hormone-refractory prostate cancer (HRPC), for which there is currently no eff ective treatment. Patients received a daily dose of 560 mg ECT in combination with 1.2 g/m2 IFM on days 1 to 5 and 70 mg/m2 CDDP on day 1. This combination therapy was given every 3 to 4 weeks. An objective response of more than 50オ reduc- tion in prostate-specifi c antigen was observed in 9 of 18 patients (50オ), and a more than 50オ reduc- tion in bi-dimensionally measurable soft-tissue lesions was observed in 2 of 7 patients (29オ). The median duration of response among the cases showing partial response was 40 weeks, while the median duration of response of overall partial-response plus stable cases was 30 weeks. The median survival duration of all cases was 47 weeks. Toxicity was modest and acceptable. In conclusion, the ECT, IFM and CDDP combination chemotherapy regimen is a viable treatment option for HRPC. -

Vinorelbine Inj. USP
VINORELBINE INJECTION USP survival between the 2 treatment groups. Survival (Figure 1) for patients receiving Vinorelbine Injection USP pIus cisplatin was Patients treated with Vinorelbine Injection USP should be frequently monitored for myelosuppression both during and after significantly better compared to the-patients who received single-agent cisplatin. The results of this trial are summarized in Table 1. therapy. Granulocytopenia is dose-limiting. Granulocyte nadirs occur between 7 and 10 days after dosing with granulocyte count PRESCRIBING INFORMATION Vinorelbine Injection USP plus Cisplatin versus Vindesine plus Cisplatin versus Single-Agent Vinorelbine Injection USP: In a recovery usually within the following 7 to 14 days. Complete blood counts with differentials should be performed and results large European clinical trial, 612 patients with Stage III or IV NSCLC, no prior chemotherapy, and WHO Performance Status of 0, 1, reviewed prior to administering each dose of Vinorelbine Injection USP. Vinorelbine Injection USP should not be administered to WARNING: Vinorelbine Injection USP should be administered under the supervision of a physician experienced in the use of or 2 were randomized to treatment with single-agent Vinorelbine Injection USP (30 mg/m2/week), Vinorelbine Injection USP (30 patients with granulocyte counts <1,000 cells/mm3. Patients developing severe granulocytopenia should be monitored carefully for cancer chemotherapeutic agents. This product is for intravenous (IV) use only. Intrathecal administration of other vinca alkaloids mg/m2/week) plus cisplatin (120 mg/m2 days 1 and 29, then every 6 weeks), and vindesine (3 mg/m2/week for 7 weeks, then every evidence of infection and/or fever. See DOSAGE AND ADMINISTRATION for recommended dose adjustments for granulocytopenia. -

Prospective Comparison of Adriamycin, Vinblastine, And
Yale University EliScholar – A Digital Platform for Scholarly Publishing at Yale Yale Medicine Thesis Digital Library School of Medicine 1988 Prospective comparison of adriamycin, vinblastine, and mitomycin C (AVM) versus adriamycin alone in the treatment of metastatic breast cancer Cristina Bengoa-Becerra Yale University Follow this and additional works at: http://elischolar.library.yale.edu/ymtdl Recommended Citation Bengoa-Becerra, Cristina, "Prospective comparison of adriamycin, vinblastine, and mitomycin C (AVM) versus adriamycin alone in the treatment of metastatic breast cancer" (1988). Yale Medicine Thesis Digital Library. 2393. http://elischolar.library.yale.edu/ymtdl/2393 This Open Access Thesis is brought to you for free and open access by the School of Medicine at EliScholar – A Digital Platform for Scholarly Publishing at Yale. It has been accepted for inclusion in Yale Medicine Thesis Digital Library by an authorized administrator of EliScholar – A Digital Platform for Scholarly Publishing at Yale. For more information, please contact [email protected]. YALE MEDICAL LIBRARY W1 ED'GAL liurtnaY .m 241988 Digitized by the Internet Archive in 2017 with funding from The National Endowment for the Humanities and the Arcadia Fund https://archive.org/details/prospectivecompaOObeng > PROSPECTIVE COMPARISON OF ADRIAMYCIN, VINBLASTINE, AND MITOMYCIN C (AVM) VERSUS ADRIAMYCIN ALONE IN THE TREATMENT OF METASTATIC BREAST CANCER A Thesis Submitted to the Yale University School of Medicine in Partial Fulfillment of the Requirements for the Degree of Doctor of Medicine by Cristina Bengoa-Becerra i 1 / 1988 - ABSTRACT PROSPECTIVE COMPARISON OF ADRIAMYCIN, VINBLASTINE, AND MITOMYCIN C (AVM) VERSUS ADR IAMYCIN ALONE IN THE TREATMENT OF METASTATIC BREAST CANCER Cristina Bengoa-Becerra 1988 Fifty-four patients with metastatic breast carcinoma refractory to conventional chemotherapy entered this prospective and randomized study.