Vinorelbine Is a Semi-Synthetic Vinca-Alkaloid with a Broad Spec- Molecular Mechanisms of Action Trum of Anti-Tumour Activity
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Cyclophosphamide-Etoposide PO Ver
Chemotherapy Protocol LYMPHOMA CYCLOPHOSPHAMIDE-ETOPOSIDE ORAL Regimen Lymphoma – Cyclophosphamide-Etoposide PO Indication Palliative treatment of malignant lymphoma Toxicity Drug Adverse Effect Cyclophosphamide Dysuria, haemorrragic cystitis (rare), taste disturbances Etoposide Alopecia, hyperbilirubinaemia The adverse effects listed are not exhaustive. Please refer to the relevant Summary of Product Characteristics for full details. Patients diagnosed with Hodgkin’s Lymphoma carry a lifelong risk of transfusion associated graft versus host disease (TA-GVHD). Where blood products are required these patients must receive only irradiated blood products for life. Local blood transfusion departments must be notified as soon as a diagnosis is made and the patient must be issued with an alert card to carry with them at all times. Monitoring Drugs FBC, LFTs and U&Es prior to day one of treatment Albumin prior to each cycle Dose Modifications The dose modifications listed are for haematological, liver and renal function and drug specific toxicities only. Dose adjustments may be necessary for other toxicities as well. In principle all dose reductions due to adverse drug reactions should not be re-escalated in subsequent cycles without consultant approval. It is also a general rule for chemotherapy that if a third dose reduction is necessary treatment should be stopped. Please discuss all dose reductions / delays with the relevant consultant before prescribing, if appropriate. The approach may be different depending on the clinical circumstances. Version 1.1 (Jan 2015) Page 1 of 6 Lymphoma- Cyclophosphamide-Etoposide PO Haematological Dose modifications for haematological toxicity in the table below are for general guidance only. Always refer to the responsible consultant as any dose reductions or delays will be dependent on clinical circumstances and treatment intent. -

Modifications on the Basic Skeletons of Vinblastine and Vincristine
Molecules 2012, 17, 5893-5914; doi:10.3390/molecules17055893 OPEN ACCESS molecules ISSN 1420-3049 www.mdpi.com/journal/molecules Review Modifications on the Basic Skeletons of Vinblastine and Vincristine Péter Keglevich, László Hazai, György Kalaus and Csaba Szántay * Department of Organic Chemistry and Technology, University of Technology and Economics, H-1111 Budapest, Szt. Gellért tér 4, Hungary * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel: +36-1-463-1195; Fax: +36-1-463-3297. Received: 30 March 2012; in revised form: 9 May 2012 / Accepted: 10 May 2012 / Published: 18 May 2012 Abstract: The synthetic investigation of biologically active natural compounds serves two main purposes: (i) the total synthesis of alkaloids and their analogues; (ii) modification of the structures for producing more selective, more effective, or less toxic derivatives. In the chemistry of dimeric Vinca alkaloids enormous efforts have been directed towards synthesizing new derivatives of the antitumor agents vinblastine and vincristine so as to obtain novel compounds with improved therapeutic properties. Keywords: antitumor therapy; vinblastine; vincristine; derivatives 1. Introduction Vinblastine (1) and vincristine (2) are dimeric alkaloids (Figure 1) isolated from the Madagaskar periwinkle plant (Catharantus roseus), exhibit significant cytotoxic activity and are used in the antitumor therapy as antineoplastic agents. In the course of cell proliferation they act as inhibitors during the metaphase of the cell cycle and by binding to the microtubules inhibit the development of the mitotic spindle. In tumor cells these agents inhibit the DNA repair and the RNA synthesis mechanisms, blocking the DNA-dependent RNA polymerase. Molecules 2012, 17 5894 Figure 1. -
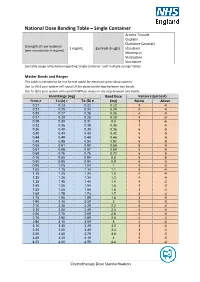
National Dose Banding Table – Single Container
National Dose Banding Table – Single Container Arsenic Trioxide Cisplatin Cladribine (Leustat) Strength of raw material 1 mg/mL Example drug(s) Idarubicin (after reconstitution if required) Mitomycin Vinblastine Vincristine See table usage notes below regarding ‘single container’ and ‘multiple syringe’ tables. Master Bands and Ranges This table is intended to be in a format useful for electronic prescribing systems. Use To (A) if your system will round UP for doses on the step between two bands. Use To (B) if your system will round DOWN for doses on the step between two bands. Band Range (mg) Band Dose Variance (percent) From ≥ To (A) < To (B) ≤ (mg) Below Above 0.21 0.23 0.22 0.22 5 -4 0.23 0.25 0.24 0.24 4 -4 0.25 0.27 0.26 0.26 4 -4 0.27 0.29 0.28 0.28 4 -3 0.29 0.32 0.31 0.3 3 -6 0.32 0.36 0.35 0.34 7 -5 0.36 0.40 0.39 0.38 6 -5 0.40 0.44 0.43 0.42 5 -5 0.44 0.49 0.48 0.46 5 -6 0.49 0.55 0.54 0.52 6 -5 0.55 0.61 0.60 0.58 6 -5 0.61 0.68 0.67 0.64 5 -6 0.68 0.76 0.75 0.72 6 -5 0.76 0.85 0.84 0.8 5 -6 0.85 0.95 0.94 0.9 6 -5 0.95 1.05 1.04 1 5 -5 1.05 1.15 1.14 1.1 5 -4 1.15 1.25 1.24 1.2 4 -4 1.25 1.35 1.34 1.3 4 -4 1.35 1.45 1.44 1.4 4 -3 1.45 1.55 1.54 1.5 4 -3 1.55 1.65 1.64 1.6 3 -3 1.65 1.75 1.74 1.7 3 -3 1.75 1.90 1.89 1.8 3 -5 1.90 2.10 2.09 2 5 -5 2.10 2.30 2.29 2.2 5 -4 2.30 2.50 2.49 2.4 4 -4 2.50 2.70 2.69 2.6 4 -4 2.70 2.90 2.89 2.8 4 -3 2.90 3.10 3.09 3 4 -3 3.10 3.30 3.29 3.2 3 -3 3.30 3.50 3.49 3.4 3 -3 3.50 3.80 3.79 3.6 3 -5 3.80 4.20 4.19 4 5 -5 4.20 4.60 4.59 4.4 5 -4 Chemotherapy Dose Standardisation Band Range (mg) -

Hodgkin Lymphoma Treatment Regimens
HODGKIN LYMPHOMA TREATMENT REGIMENS (Part 1 of 5) Clinical Trials: The National Comprehensive Cancer Network recommends cancer patient participation in clinical trials as the gold standard for treatment. Cancer therapy selection, dosing, administration, and the management of related adverse events can be a complex process that should be handled by an experienced health care team. Clinicians must choose and verify treatment options based on the individual patient; drug dose modifications and supportive care interventions should be administered accordingly. The cancer treatment regimens below may include both U.S. Food and Drug Administration-approved and unapproved indications/regimens. These regimens are provided only to supplement the latest treatment strategies. These Guidelines are a work in progress that may be refined as often as new significant data become available. The NCCN Guidelines® are a consensus statement of its authors regarding their views of currently accepted approaches to treatment. Any clinician seeking to apply or consult any NCCN Guidelines® is expected to use independent medical judgment in the context of individual clinical circumstances to determine any patient’s care or treatment. The NCCN makes no warranties of any kind whatsoever regarding their content, use, or application and disclaims any responsibility for their application or use in any way. Classical Hodgkin Lymphoma1 Note: All recommendations are Category 2A unless otherwise indicated. Primary Treatment Stage IA, IIA Favorable (No Bulky Disease, <3 Sites of Disease, ESR <50, and No E-lesions) REGIMEN DOSING Doxorubicin + Bleomycin + Days 1 and 15: Doxorubicin 25mg/m2 IV push + bleomycin 10units/m2 IV push + Vinblastine + Dacarbazine vinblastine 6mg/m2 IV over 5–10 minutes + dacarbazine 375mg/m2 IV over (ABVD) (Category 1)2-5 60 minutes. -

Vincristine (Conventional): Drug Information
Official reprint from UpToDate® www.uptodate.com ©2017 UpToDate® Vincristine (conventional): Drug information Copyright 1978-2017 Lexicomp, Inc. All rights reserved. (For additional information see "Vincristine (conventional): Patient drug information" and see "Vincristine (conventional): Pediatric drug information") For abbreviations and symbols that may be used in Lexicomp (show table) Special Alerts Vincristine Sulfate Safety Alert October 2015 Health Canada is notifying health care providers that certain lots of Hospira’s vincristine sulfate 1 mg/mL injection (DIN 02183013: 2 mL vial, list #7077A001; 5 mL vial, list #7082A001) have incorrect or outdated safety information on the inner/outer labels and package insert, which may increase the risk to patients and may result in significant patient harm requiring medical intervention. These warnings include: - Vincristine should only be administered by the intravenous (IV) route. Administration of vincristine by any other route can be fatal. - Syringes containing this product should be labeled “Warning - for IV use only.” - Extemporaneously prepared syringes containing this product must be packaged in an overwrap which is labeled “Do not remove covering until moment of injection. For IV use only - fatal if given by other routes.” - Contraindication of vincristine in patients with demyelinating Charcot-Marie-Tooth syndrome. - Potential risk of acute shortness of breath when vincristine is coadministered with mitomycin-C and GI toxicities including necrosis with administration of vincristine. Health care providers are requested to consult with the approved Canadian product monograph for vincristine sulfate 1 mg/mL for the most updated information. Consumers with questions should contact their health care provider for more information. ALERT: US Boxed Warning Experienced physician: Vincristine should be administered by individuals experienced in the administration of the drug. -

BC Cancer Protocol Summary for Treatment of Lymphoma with Dose- Adjusted Etoposide, Doxorubicin, Vincristine, Cyclophosphamide
BC Cancer Protocol Summary for Treatment of Lymphoma with Dose- Adjusted Etoposide, DOXOrubicin, vinCRIStine, Cyclophosphamide, predniSONE and riTUXimab with Intrathecal Methotrexate Protocol Code LYEPOCHR Tumour Group Lymphoma Contact Physician Dr. Laurie Sehn Dr. Kerry Savage ELIGIBILITY: One of the following lymphomas: . Patients with an aggressive B-cell lymphoma and the presence of a dual translocation of MYC and BCL2 (i.e., double-hit lymphoma). Histologies may include DLBCL, transformed lymphoma, unclassifiable lymphoma, and intermediate grade lymphoma, not otherwise specified (NOS). Patients with Burkitt lymphoma, who are not candidates for CODOXM/IVACR (such as those over the age of 65 years, or with significant co-morbidities) . Primary mediastinal B-cell lymphoma Ensure patient has central line EXCLUSIONS: . Cardiac dysfunction that would preclude the use of an anthracycline. TESTS: . Baseline (required before first treatment): CBC and diff, platelets, BUN, creatinine, bilirubin. ALT, LDH, uric acid . Baseline (required, but results do not have to be available to proceed with first treatment): results must be checked before proceeding with cycle 2): HBsAg, HBcoreAb, . Baseline (optional, results do not have to be available to proceed with first treatment): HCAb, HIV . Day 1 of each cycle: CBC and diff, platelets, (and serum bilirubin if elevated at baseline; serum bilirubin does not need to be requested before each treatment, after it has returned to normal), urinalysis for microscopic hematuria (optional) . Days 2 and 5 of each cycle (or days of intrathecal treatment): CBC and diff, platelets, PTT, INR . For patients on cyclophosphamide doses greater than 2000 mg: Daily urine dipstick for blood starting on day cyclophosphamide is given. -

In Relation to HIFU Treatment on the Growth of Dunning Tumors: Results of a Preliminary Study
Prostate Cancer and Prostatic Diseases (2008) 11, 181–186 & 2008 Nature Publishing Group All rights reserved 1365-7852/08 $30.00 www.nature.com/pcan ORIGINAL ARTICLE Influence of the docetaxel administration period (neoadjuvant or concomitant) in relation to HIFU treatment on the growth of Dunning tumors: results of a preliminary study P Paparel1,2, JY Chapelon2, A Bissery3, S Chesnais2, L Curiel2 and A Gelet2,4 1Department of Urology, Lyon Sud Hospital, Pierre Be´nite, France; 2INSERM U 556, Lyon, France; 3Department of Biostatistics, Hospices civils de Lyon, Lyon, France and 4Department of Urology, Edouard Herriot Hospital, Lyon, France The objective of this study was to evaluate mechanisms of the synergy between high intensity- focused ultrasound (HIFU) and docetaxel and to determine the best sequence of chemotherapy administration in relation to HIFU treatment for obtaining optimum control of tumoral growth. A total of 15 days after s.c. implantation of the tumor, 52 Copenhagen rats studied were randomized in 4 groups of 13: controls, docetaxel alone (group 1), HIFU and docetaxel concomitant (group 2) and HIFU and docetaxel administered 24 h before treatment (group 3). The number of HIFU shots was calculated in order to cover 75% of the tumor volume. The effects of docetaxel, HIFU and their interaction on tumor volumes were analyzed using a linear regression. The distributions of the tumor volumes were significantly greater in the control group than in the group 1 (P ¼ 0.002) and than in both groups 2 and 3 (Po0.0001 and P ¼ 0.0001). These volumes were also significantly greater in group 1 than in both groups 2 and 3 and there was no difference between the groups 2 and 3. -

Arsenic Trioxide Is Highly Cytotoxic to Small Cell Lung Carcinoma Cells
160 Arsenic trioxide is highly cytotoxic to small cell lung carcinoma cells 1 1 Helen M. Pettersson, Alexander Pietras, effect of As2O3 on SCLC growth, as suggested by an Matilda Munksgaard Persson,1 Jenny Karlsson,1 increase in neuroendocrine markers in cultured cells. [Mol Leif Johansson,2 Maria C. Shoshan,3 Cancer Ther 2009;8(1):160–70] and Sven Pa˚hlman1 1Center for Molecular Pathology, CREATE Health and 2Division of Introduction Pathology, Department of Laboratory Medicine, Lund University, 3 Lung cancer is the most frequent cause of cancer deaths University Hospital MAS, Malmo¨, Sweden; and Department of f Oncology-Pathology, Cancer Center Karolinska, Karolinska worldwide and results in 1 million deaths each year (1). Institute and Hospital, Stockholm, Sweden Despite novel treatment strategies, the 5-year survival rate of lung cancer patients is only f15%. Small cell lung carcinoma (SCLC) accounts for 15% to 20% of all lung Abstract cancers diagnosed and is a very aggressive malignancy Small cell lung carcinoma (SCLC) is an extremely with early metastatic spread (2). Despite an initially high aggressive form of cancer and current treatment protocols rate of response to chemotherapy, which currently com- are insufficient. SCLC have neuroendocrine characteristics bines a platinum-based drug with another cytotoxic drug and show phenotypical similarities to the childhood tumor (3, 4), relapses occur in the absolute majority of SCLC neuroblastoma. As multidrug-resistant neuroblastoma patients. At relapse, the efficacy of further chemotherapy is cells are highly sensitive to arsenic trioxide (As2O3) poor and the need for alternative treatments is obvious. in vitro and in vivo, we here studied the cytotoxic effects Arsenic-containing compounds have been used in tradi- of As2O3 on SCLC cells. -

Chemotherapy Protocol
Chemotherapy Protocol BREAST CANCER CYCLOPHOSPHAMIDE-EPIRUBICIN-PACLITAXEL (7 day) Regimen • Breast Cancer – Cyclophosphamide-Epirubicin-Paclitaxel (7 day) Indication • Neoadjuvant / adjuvant therapy of early breast cancer • WHO Performance status 0, 1 Toxicity Drug Adverse Effect Cyclophosphamide Dysuria, haemorrhagic cystitis, taste disturbances Epirubicin Cardio-toxicity, urinary discolouration (red) Hypersensitivity, hypotension, bradycardia, peripheral Paclitaxel neuropathy, myalgia and back pain on administration The adverse effects listed are not exhaustive. Please refer to the relevant Summary of Product Characteristics for full details. Monitoring Regimen • FBC, U&Es and LFTs prior to each cycle. • Ensure adequate cardiac function before starting treatment with epirubicin. Baseline LVEF should be measured, particularly in patients with a history of cardiac problems or in the elderly. Dose Modifications The dose modifications listed are for haematological, liver and renal function only. Dose adjustments may be necessary for other toxicities as well. In principle all dose reductions due to adverse drug reactions should not be re- escalated in subsequent cycles without consultant approval. It is also a general rule for chemotherapy that if a third dose reduction is necessary treatment should be stopped. Please discuss all dose reductions / delays with the relevant consultant before prescribing if appropriate. The approach may be different depending on the clinical circumstances. The following is a general guide only. Version 1.2 (November 2020) Page 1 of 7 Breast – Cyclophosphamide-Epirubicin-Paclitaxel (7 day) Haematological Prior to prescribing the following treatment criteria must be met on day one of treatment. Criteria Eligible Level Neutrophils equal to or more than 1x10 9/L Platelets equal to or more than 100x10 9/L Consider blood transfusion if patient symptomatic of anaemia or has a haemoglobin of less than 8g/dL If counts on day one are below these criteria for neutrophils and platelets then delay treatment for seven days. -

Ethylene-Induced Vinblastine Accumulation Is Related to Activated Expression of Downstream TIA Pathway Genes in Catharanthus Roseus
Hindawi Publishing Corporation BioMed Research International Volume 2016, Article ID 3708187, 8 pages http://dx.doi.org/10.1155/2016/3708187 Research Article Ethylene-Induced Vinblastine Accumulation Is Related to Activated Expression of Downstream TIA Pathway Genes in Catharanthus roseus Xi Wang,1 Ya-Jie Pan,1 Bo-Wen Chang,2 Yan-Bo Hu,2 Xiao-Rui Guo,1 and Zhong-Hua Tang1 1 Key Laboratory of Forest Plant Ecology, Northeast Forestry University, Harbin 150040, China 2College of Life Science, Northeast Forestry University, Harbin 150040, China Correspondence should be addressed to Xiao-Rui Guo; [email protected] and Zhong-Hua Tang; [email protected] Received 13 December 2015; Revised 30 March 2016; Accepted 6 April 2016 Academic Editor: Sudhir Sopory Copyright © 2016 Xi Wang et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. We selected different concentrations of ethephon, to stress C. roseus.WeusedqRT-PCRandHPLCfollowedbyPCAtoobtain comprehensive profiling of the vinblastine biosynthesis in response to ethephon. Based on our findings, the results showed thatthe high concentration of ethephon had a positive effect at both transcriptional and metabolite level. Meanwhile, there was a remarkable decrease of hydrogen peroxide content and a promoted peroxidase activity in leaves. The loading plot combination with correlation analysis suggested that CrPrx1 could be regarded as a positive regulator and interacts with ethylene response factor (ERF)toplaya key role in vinblastine content and peroxidase (POD) activity. This study provides the foundation for a better understanding of the regulation and accumulation of vinblastine in response to ethephon. -

The Iboga Alkaloids
The Iboga Alkaloids Catherine Lavaud and Georges Massiot Contents 1 Introduction ................................................................................. 90 2 Biosynthesis ................................................................................. 92 3 Structural Elucidation and Reactivity ...................................................... 93 4 New Molecules .............................................................................. 97 4.1 Monomers ............................................................................. 99 4.1.1 Ibogamine and Coronaridine Derivatives .................................... 99 4.1.2 3-Alkyl- or 3-Oxo-ibogamine/-coronaridine Derivatives . 102 4.1.3 5- and/or 6-Oxo-ibogamine/-coronaridine Derivatives ...................... 104 4.1.4 Rearranged Ibogamine/Coronaridine Alkaloids .. ........................... 105 4.1.5 Catharanthine and Pseudoeburnamonine Derivatives .. .. .. ... .. ... .. .. ... .. 106 4.1.6 Miscellaneous Representatives and Another Enigma . ..................... 107 4.2 Dimers ................................................................................. 108 4.2.1 Bisindoles with an Ibogamine Moiety ....................................... 110 4.2.2 Bisindoles with a Voacangine (10-Methoxy-coronaridine) Moiety ........ 111 4.2.3 Bisindoles with an Isovoacangine (11-Methoxy-coronaridine) Moiety . 111 4.2.4 Bisindoles with an Iboga-Indolenine or Rearranged Moiety ................ 116 4.2.5 Bisindoles with a Chippiine Moiety ... ..................................... -

Metals and Metal Compounds in Cancer Treatment
ANTICANCER RESEARCH 24: 1529-1544 (2004) Review Metals and Metal Compounds in Cancer Treatment BERNARD DESOIZE Laboratoire de Biochimie et de Biologie Moléculaire, EA 3306, IFR 53, Faculté de Pharmacie, 51 rue Cognacq-Jay, 51096 Reims cedex, France Abstract. Metals and metal compounds have been used in The importance of metal compounds in medicine is medicine for several thousands of years. In this review we undisputed, as can be judged by the use of compounds of summarized the anti-cancer activities of the ten most active antimony (anti-protozoal), bismuth (anti-ulcer), gold (anti- metals: arsenic, antimony, bismuth, gold, vanadium, iron, arthritic), iron (anti-malarial), silver (anti-microbial) and rhodium, titanium, gallium and platinum. The first reviewed platinum (anti-cancer) in the treatment of various diseases. metal, arsenic, presents the anomaly of displaying anti-cancer In terms of anti-tumour activity, a wide range of compounds and oncogenic properties simultaneously. Some antimony of both transition metals and main group elements have derivatives, such as Sb2O3, salt (tartrate) and organic been investigated for efficacy (1). The earliest reports on the compounds, show interesting results. Bismuth directly affects therapeutic use of metals or metal-containing compounds in Helicobacter pylori and gastric lymphoma; the effects of cancer and leukemia date from the sixteenth century. bismuth complexes of 6-mercaptopurine are promising. Superoxide dismutase inhibition leads to selective killing Gold(I) and (III) compounds show anti-tumour activities, of cancer cells in vitro and in vivo (2,3). As a consequence, although toxicity remains high. Research into the potential use reactive oxygen species (ROS) may not only be involved in of gold derivatives is still ongoing.