Modifications on the Basic Skeletons of Vinblastine and Vincristine
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Carbamate Pesticides Aldicarb Aldicarb Sulfoxide Aldicarb Sulfone
Connecticut General Statutes Sec 19a-29a requires the Commissioner of Public Health to annually publish a list setting forth all analytes and matrices for which certification for testing is required. Connecticut ELCP Drinking Water Analytes Revised 05/31/2018 Microbiology Total Coliforms Fecal Coliforms/ E. Coli Carbamate Pesticides Legionella Aldicarb Cryptosporidium Aldicarb Sulfoxide Giardia Aldicarb Sulfone Carbaryl Physicals Carbofuran Turbidity 3-Hydroxycarbofuran pH Methomyl Conductivity Oxamyl (Vydate) Minerals Chlorinated Herbicides Alkalinity, as CaCO3 2,4-D Bromide Dalapon Chloride Dicamba Chlorine, free residual Dinoseb Chlorine, total residual Endothall Fluoride Picloram Hardness, Calcium as Pentachlorophenol CaCO3 Hardness, Total as CaCO3 Silica Chlorinated Pesticides/PCB's Sulfate Aldrin Chlordane (Technical) Nutrients Dieldrin Endrin Ammonia Heptachlor Nitrate Heptachlor Epoxide Nitrite Lindane (gamma-BHC) o-Phosphate Metolachlor Total Phosphorus Methoxychlor PCB's (individual aroclors) Note 1 PCB's (as decachlorobiphenyl) Note 1 Demands Toxaphene TOC Nitrogen-Phosphorus Compounds Alachlor Metals Atrazine Aluminum Butachlor Antimony Diquat Arsenic Glyphosate Barium Metribuzin Beryllium Paraquat Boron Propachlor Cadmium Simazine Calcium Chromium Copper SVOC's Iron Benzo(a)pyrene Lead bis-(2-ethylhexyl)phthalate Magnesium bis-(ethylhexyl)adipate Manganese Hexachlorobenzene Mercury Hexachlorocyclopentadiene Molybdenum Nickel Potassium Miscellaneous Organics Selenium Dibromochloropropane (DBCP) Silver Ethylene Dibromide (EDB) -
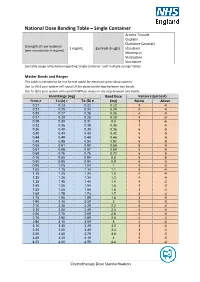
National Dose Banding Table – Single Container
National Dose Banding Table – Single Container Arsenic Trioxide Cisplatin Cladribine (Leustat) Strength of raw material 1 mg/mL Example drug(s) Idarubicin (after reconstitution if required) Mitomycin Vinblastine Vincristine See table usage notes below regarding ‘single container’ and ‘multiple syringe’ tables. Master Bands and Ranges This table is intended to be in a format useful for electronic prescribing systems. Use To (A) if your system will round UP for doses on the step between two bands. Use To (B) if your system will round DOWN for doses on the step between two bands. Band Range (mg) Band Dose Variance (percent) From ≥ To (A) < To (B) ≤ (mg) Below Above 0.21 0.23 0.22 0.22 5 -4 0.23 0.25 0.24 0.24 4 -4 0.25 0.27 0.26 0.26 4 -4 0.27 0.29 0.28 0.28 4 -3 0.29 0.32 0.31 0.3 3 -6 0.32 0.36 0.35 0.34 7 -5 0.36 0.40 0.39 0.38 6 -5 0.40 0.44 0.43 0.42 5 -5 0.44 0.49 0.48 0.46 5 -6 0.49 0.55 0.54 0.52 6 -5 0.55 0.61 0.60 0.58 6 -5 0.61 0.68 0.67 0.64 5 -6 0.68 0.76 0.75 0.72 6 -5 0.76 0.85 0.84 0.8 5 -6 0.85 0.95 0.94 0.9 6 -5 0.95 1.05 1.04 1 5 -5 1.05 1.15 1.14 1.1 5 -4 1.15 1.25 1.24 1.2 4 -4 1.25 1.35 1.34 1.3 4 -4 1.35 1.45 1.44 1.4 4 -3 1.45 1.55 1.54 1.5 4 -3 1.55 1.65 1.64 1.6 3 -3 1.65 1.75 1.74 1.7 3 -3 1.75 1.90 1.89 1.8 3 -5 1.90 2.10 2.09 2 5 -5 2.10 2.30 2.29 2.2 5 -4 2.30 2.50 2.49 2.4 4 -4 2.50 2.70 2.69 2.6 4 -4 2.70 2.90 2.89 2.8 4 -3 2.90 3.10 3.09 3 4 -3 3.10 3.30 3.29 3.2 3 -3 3.30 3.50 3.49 3.4 3 -3 3.50 3.80 3.79 3.6 3 -5 3.80 4.20 4.19 4 5 -5 4.20 4.60 4.59 4.4 5 -4 Chemotherapy Dose Standardisation Band Range (mg) -

Hodgkin Lymphoma Treatment Regimens
HODGKIN LYMPHOMA TREATMENT REGIMENS (Part 1 of 5) Clinical Trials: The National Comprehensive Cancer Network recommends cancer patient participation in clinical trials as the gold standard for treatment. Cancer therapy selection, dosing, administration, and the management of related adverse events can be a complex process that should be handled by an experienced health care team. Clinicians must choose and verify treatment options based on the individual patient; drug dose modifications and supportive care interventions should be administered accordingly. The cancer treatment regimens below may include both U.S. Food and Drug Administration-approved and unapproved indications/regimens. These regimens are provided only to supplement the latest treatment strategies. These Guidelines are a work in progress that may be refined as often as new significant data become available. The NCCN Guidelines® are a consensus statement of its authors regarding their views of currently accepted approaches to treatment. Any clinician seeking to apply or consult any NCCN Guidelines® is expected to use independent medical judgment in the context of individual clinical circumstances to determine any patient’s care or treatment. The NCCN makes no warranties of any kind whatsoever regarding their content, use, or application and disclaims any responsibility for their application or use in any way. Classical Hodgkin Lymphoma1 Note: All recommendations are Category 2A unless otherwise indicated. Primary Treatment Stage IA, IIA Favorable (No Bulky Disease, <3 Sites of Disease, ESR <50, and No E-lesions) REGIMEN DOSING Doxorubicin + Bleomycin + Days 1 and 15: Doxorubicin 25mg/m2 IV push + bleomycin 10units/m2 IV push + Vinblastine + Dacarbazine vinblastine 6mg/m2 IV over 5–10 minutes + dacarbazine 375mg/m2 IV over (ABVD) (Category 1)2-5 60 minutes. -

Mandatory Standard for Vinca Alkaloids
Mandatory Standard for vinca alkaloids 1. Introduction Vinca alkaloids are a class of chemotherapeutic agents which includes vinblastine, vincristine, vinflunine and vinorelbine. Health Service Providers are required to have local guidelines in place which adhere to the minimum safety requirements outlined in this Standard. This Standard is based on a national alert on vinca alkaloids developed by the Australian Council for Safety and Quality in Health Care. The Australian Commission for Safety and Quality in Health Care published an updated alert in February 2019.1 2. Vinca Alkaloids Standard While any medication administered via the incorrect route can result in harm, the inadvertent intrathecal administration of vinca alkaloids has resulted in death or permanent disability and remains a potential risk.2 Vinca alkaloids can be fatal if given by the intrathecal route and must therefore ONLY be administered intravenously. Specific requirements for hospitals and health services are outlined below. 2.1 Prescribing requirements • In accordance with the Clinical Oncological Society of Australia guidelines, vinca alkaloids must only be prescribed by medical practitioners with appropriate skills, training and qualifications in the management of cancer. • Dosing of vinca alkaloids must be calculated by a medical practitioner skilled in this task. 2.2 Preparation and dispensing • All vinca alkaloids must be prepared and supplied in a minibag of compatible solution, never in a syringe. For adult dosing the minibag must contain a total volume of 50mL or more. • Vinca alkaloids must only be prepared and dispensed by appropriately trained staff that have been assessed as competent to prepare and dispense chemotherapy. The total milligram dose of vinca alkaloid to be added to the minibag must be verified by a chemotherapy competent pharmacist before it is dispensed in the Pharmacy Compounding Unit. -

WO 2018/005077 Al O O© O
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2018/005077 Al 04 January 2018 (04.01.2018) W ! P O PCT (51) International Patent Classification: SC, SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, A61K 31/78 (2006.01) C08J 7/04 (2006.01) TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. C08G 59/77 (2006.01) (84) Designated States (unless otherwise indicated, for every (21) International Application Number: kind of regional protection available): ARIPO (BW, GH, PCT/US20 17/037 176 GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, ST, SZ, TZ, UG, ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, RU, TJ, (22) International Filing Date: TM), European (AL, AT, BE, BG, CH, CY, CZ, DE, DK, 13 June 2017 (13.06.2017) EE, ES, FI, FR, GB, GR, HR, HU, IE, IS, IT, LT, LU, LV, (25) Filing Language: English MC, MK, MT, NL, NO, PL, PT, RO, RS, SE, SI, SK, SM, TR), OAPI (BF, BJ, CF, CG, CI, CM, GA, GN, GQ, GW, (26) Publication Language: English KM, ML, MR, NE, SN, TD, TG). (30) Priority Data: 62/356,918 30 June 2016 (30.06.2016) US Published: — with international search report (Art. 21(3)) (71) Applicant: ELEMENTIS SPECIALTIES, INC. [US/US]; 469 Old Trenton Road, East Windsor, NJ 085 12 (US). (72) Inventors: IJDO, Wouter; 1224 Bridle Estates Dri ve, Yardley, PA 19067 (US). CHEN, Yanhui; 4 Hal- stead Place, Princeton, NJ 08540 (US). -

Ethylene-Induced Vinblastine Accumulation Is Related to Activated Expression of Downstream TIA Pathway Genes in Catharanthus Roseus
Hindawi Publishing Corporation BioMed Research International Volume 2016, Article ID 3708187, 8 pages http://dx.doi.org/10.1155/2016/3708187 Research Article Ethylene-Induced Vinblastine Accumulation Is Related to Activated Expression of Downstream TIA Pathway Genes in Catharanthus roseus Xi Wang,1 Ya-Jie Pan,1 Bo-Wen Chang,2 Yan-Bo Hu,2 Xiao-Rui Guo,1 and Zhong-Hua Tang1 1 Key Laboratory of Forest Plant Ecology, Northeast Forestry University, Harbin 150040, China 2College of Life Science, Northeast Forestry University, Harbin 150040, China Correspondence should be addressed to Xiao-Rui Guo; [email protected] and Zhong-Hua Tang; [email protected] Received 13 December 2015; Revised 30 March 2016; Accepted 6 April 2016 Academic Editor: Sudhir Sopory Copyright © 2016 Xi Wang et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. We selected different concentrations of ethephon, to stress C. roseus.WeusedqRT-PCRandHPLCfollowedbyPCAtoobtain comprehensive profiling of the vinblastine biosynthesis in response to ethephon. Based on our findings, the results showed thatthe high concentration of ethephon had a positive effect at both transcriptional and metabolite level. Meanwhile, there was a remarkable decrease of hydrogen peroxide content and a promoted peroxidase activity in leaves. The loading plot combination with correlation analysis suggested that CrPrx1 could be regarded as a positive regulator and interacts with ethylene response factor (ERF)toplaya key role in vinblastine content and peroxidase (POD) activity. This study provides the foundation for a better understanding of the regulation and accumulation of vinblastine in response to ethephon. -

The Iboga Alkaloids
The Iboga Alkaloids Catherine Lavaud and Georges Massiot Contents 1 Introduction ................................................................................. 90 2 Biosynthesis ................................................................................. 92 3 Structural Elucidation and Reactivity ...................................................... 93 4 New Molecules .............................................................................. 97 4.1 Monomers ............................................................................. 99 4.1.1 Ibogamine and Coronaridine Derivatives .................................... 99 4.1.2 3-Alkyl- or 3-Oxo-ibogamine/-coronaridine Derivatives . 102 4.1.3 5- and/or 6-Oxo-ibogamine/-coronaridine Derivatives ...................... 104 4.1.4 Rearranged Ibogamine/Coronaridine Alkaloids .. ........................... 105 4.1.5 Catharanthine and Pseudoeburnamonine Derivatives .. .. .. ... .. ... .. .. ... .. 106 4.1.6 Miscellaneous Representatives and Another Enigma . ..................... 107 4.2 Dimers ................................................................................. 108 4.2.1 Bisindoles with an Ibogamine Moiety ....................................... 110 4.2.2 Bisindoles with a Voacangine (10-Methoxy-coronaridine) Moiety ........ 111 4.2.3 Bisindoles with an Isovoacangine (11-Methoxy-coronaridine) Moiety . 111 4.2.4 Bisindoles with an Iboga-Indolenine or Rearranged Moiety ................ 116 4.2.5 Bisindoles with a Chippiine Moiety ... ..................................... -

The Phytochemical Analysis of Vinca L. Species Leaf Extracts Is Correlated with the Antioxidant, Antibacterial, and Antitumor Effects
molecules Article The Phytochemical Analysis of Vinca L. Species Leaf Extracts Is Correlated with the Antioxidant, Antibacterial, and Antitumor Effects 1,2, 3 3 1 1 Alexandra Ciorît, ă * , Cezara Zăgrean-Tuza , Augustin C. Mot, , Rahela Carpa and Marcel Pârvu 1 Faculty of Biology and Geology, Babes, -Bolyai University, 44 Republicii St., 400015 Cluj-Napoca, Romania; [email protected] (R.C.); [email protected] (M.P.) 2 National Institute for Research and Development of Isotopic and Molecular Technologies, 67-103 Donath St., 400293 Cluj-Napoca, Romania 3 Faculty of Chemistry and Chemical Engineering, Babes, -Bolyai University, 11 Arany János St., 400028 Cluj-Napoca, Romania; [email protected] (C.Z.-T.); [email protected] (A.C.M.) * Correspondence: [email protected]; Tel.: +40-264-584-037 Abstract: The phytochemical analysis of Vinca minor, V. herbacea, V. major, and V. major var. variegata leaf extracts showed species-dependent antioxidant, antibacterial, and cytotoxic effects correlated with the identified phytoconstituents. Vincamine was present in V. minor, V. major, and V. major var. variegata, while V. minor had the richest alkaloid content, followed by V. herbacea. V. major var. variegata was richest in flavonoids and the highest total phenolic content was found in V. herbacea which also had elevated levels of rutin. Consequently, V. herbacea had the highest antioxidant activity V. major variegata V. major V. minor followed by var. Whereas, the lowest one was of . The extract showed the most efficient inhibitory effect against both Staphylococcus aureus and E. coli. On the other hand, V. herbacea had a good anti-bacterial potential only against S. -

BC Cancer Benefit Drug List September 2021
Page 1 of 65 BC Cancer Benefit Drug List September 2021 DEFINITIONS Class I Reimbursed for active cancer or approved treatment or approved indication only. Reimbursed for approved indications only. Completion of the BC Cancer Compassionate Access Program Application (formerly Undesignated Indication Form) is necessary to Restricted Funding (R) provide the appropriate clinical information for each patient. NOTES 1. BC Cancer will reimburse, to the Communities Oncology Network hospital pharmacy, the actual acquisition cost of a Benefit Drug, up to the maximum price as determined by BC Cancer, based on the current brand and contract price. Please contact the OSCAR Hotline at 1-888-355-0355 if more information is required. 2. Not Otherwise Specified (NOS) code only applicable to Class I drugs where indicated. 3. Intrahepatic use of chemotherapy drugs is not reimbursable unless specified. 4. For queries regarding other indications not specified, please contact the BC Cancer Compassionate Access Program Office at 604.877.6000 x 6277 or [email protected] DOSAGE TUMOUR PROTOCOL DRUG APPROVED INDICATIONS CLASS NOTES FORM SITE CODES Therapy for Metastatic Castration-Sensitive Prostate Cancer using abiraterone tablet Genitourinary UGUMCSPABI* R Abiraterone and Prednisone Palliative Therapy for Metastatic Castration Resistant Prostate Cancer abiraterone tablet Genitourinary UGUPABI R Using Abiraterone and prednisone acitretin capsule Lymphoma reversal of early dysplastic and neoplastic stem changes LYNOS I first-line treatment of epidermal -

(10) Patent No.: US 9562099 B2
USO09562099B2 (12) United States Patent (10) Patent No.: US 9,562,099 B2 Leong et al. (45) Date of Patent: Feb. 7, 2017 (54) ANTI-B7-H4 ANTIBODIES AND 4,260,608 A 4, 1981 Miyashita et al. MMUNOCONUGATES 4,265,814 A 5/1981 Hashimoto et al. 4.294,757 A 10, 1981 Asai (71) Applicant: Genentech, Inc., South San Francisco, 4,307,016 A 12/1981 Asai et al. CA (US) 4,308,268 A 12/1981 Miyashita et al. 4,308,269 A 12/1981 Miyashita et al. (72) Inventors: Steven R. Leong, Berkeley, CA (US); 4,309.428 A 1/1982 Miyashita et al. Andrew Polson, San Francisco, CA 4,313,946 A 2f1982 Powell et al. (US); s Paul Polakis,s Mill Valley,y, CA 4,317,8214,315,929 A 3/19822, 1982 MiyashitaFreedman et al. (US); Yan Wu, Foster City, CA (US); 4.322,348 A 3/1982 Asai et al. Wei-Ching Liang, Foster City, CA 4.331,598. A 5/1982 Hasegawa et al. (US); Ron Firestein, Burlingame, CA 4,361,650 A 1 1/1982 Asai et al. (US) 4,362,663 A 12/1982 Kida et al. 4,364,866 A 12/1982 Asai et al. (73) Assignee: Genentech, Inc., South San Francisco, 4,371.533. A 2, 1983 Akimoto et al. CA (US) 4.424,219 A 1/1984 Hashimoto et al. 4.450,254 A 5/1984 Isley et al. (*) Notice: Subject to any disclaimer, the term of this 4,676.980 A 6/1987 Segal et al. -

N-Methyl Carbamate Cumulative Risk Assessment: Pilot Cumulative Analysis
UNITED STATES ENVIRONMENTAL PROTECTION AGENCY WASHINGTON, D.C. 20460 OFFICE OF PREVENTION, PESTICIDES, AND TOXIC SUBSTANCES April 15, 2005 MEMORANDUM SUBJECT: Transmittal of Meeting Minutes of the FIFRA Scientific Advisory Panel Meeting Held February 15 - 18, 2005 on the N-methyl Carbamate Cumulative Risk Assessment: Pilot Cumulative Analysis TO: James J. Jones, Director Office of Pesticide Programs FROM: Myrta R. Christian, Designated Federal Official Joseph E. Bailey, Designated Federal Official FIFRA Scientific Advisory Panel Office of Science Coordination and Policy THRU: Larry C. Dorsey, Executive Secretary FIFRA Scientific Advisory Panel Office of Science Coordination and Policy Clifford J. Gabriel, Director Office of Science Coordination and Policy Attached, please find the meeting minutes of the FIFRA Scientific Advisory Panel open meeting held in Arlington, Virginia on February 15 - 18, 2005. This report addresses a set of scientific issues being considered by the Environmental Protection Agency pertaining to the N- methyl carbamate cumulative risk assessment: pilot cumulative analysis. Attachment 1 of 113 cc: Susan Hazen Margaret Schneider Anne Lindsay Margie Fehrenbach Janet Andersen Debbie Edwards Steven Bradbury William Diamond Arnold Layne Tina Levine Lois Rossi Frank Sanders Richard Keigwin Randolph Perfetti William Jordan Douglas Parsons Enesta Jones Vanessa Vu (SAB) Anna Lowit David J. Miller Nelson Thurman Dirk Young David Hrdy Jeff Evans Steve Nako Stephanie Padilla R. Woodrow Setzer Ginger Moser Miles Okino Jerry Blancato Fred Power Curtis Dary Tom Nolan, USGS OPP Docket 2 of 113 FIFRA Scientific Advisory Panel Members Stephen M. Roberts, Ph.D. (Chair of the FIFRA SAP) Janice E. Chambers, Ph.D. H. Christopher Frey, Ph.D. -

Collected Mass Spectrometry Data on Monoterpene Indole Alkaloids from Natural Product Chemistry Research
www.nature.com/scientificdata OPEN Collected mass spectrometry data DATA DescrIPTOR on monoterpene indole alkaloids from natural product chemistry Received: 20 September 2018 Accepted: 25 February 2019 research Published: xx xx xxxx Alexander E. Fox Ramos 1, Pierre Le Pogam1, Charlotte Fox Alcover 1, Elvis Otogo N’Nang 1, Gaëla Cauchie 1, Hazrina Hazni1,2, Khalijah Awang2, Dimitri Bréard3, Antonio M. Echavarren4,5, Michel Frédérich6, Thomas Gaslonde7, Marion Girardot8, Raphaël Grougnet7, Mariia S. Kirillova4, Marina Kritsanida 7, Christelle Lémus7, Anne-Marie Le Ray3, Guy Lewin1, Marc Litaudon9, Lengo Mambu 10, Sylvie Michel7, Fedor M. Miloserdov4, Michael E. Muratore4, Pascal Richomme-Peniguel3, Fanny Roussi9, Laurent Evanno1, Erwan Poupon1, Pierre Champy1 & Mehdi A. Beniddir 1 This Data Descriptor announces the submission to public repositories of the monoterpene indole alkaloid database (MIADB), a cumulative collection of 172 tandem mass spectrometry (MS/MS) spectra from multiple research projects conducted in eight natural product chemistry laboratories since the 1960s. All data have been annotated and organized to promote reuse by the community. Being a unique collection of these complex natural products, these data can be used to guide the dereplication and targeting of new related monoterpene indole alkaloids within complex mixtures when applying computer-based approaches, such as molecular networking. Each spectrum has its own accession number from CCMSLIB00004679916 to CCMSLIB00004680087 on the GNPS. The MIADB is available for download from MetaboLights under the identifer: MTBLS142 (https://www.ebi.ac.uk/metabolights/ MTBLS142). Background & Summary Monoterpene indole alkaloids (MIAs) constitute a broad class of nitrogen-containing plant-derived natural products composed of more than 3000 members1.