RNA Sequencing to Explore Dominant Isoform Switch During CCR6+ Memory T Cell Activation
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

PARSANA-DISSERTATION-2020.Pdf
DECIPHERING TRANSCRIPTIONAL PATTERNS OF GENE REGULATION: A COMPUTATIONAL APPROACH by Princy Parsana A dissertation submitted to The Johns Hopkins University in conformity with the requirements for the degree of Doctor of Philosophy Baltimore, Maryland July, 2020 © 2020 Princy Parsana All rights reserved Abstract With rapid advancements in sequencing technology, we now have the ability to sequence the entire human genome, and to quantify expression of tens of thousands of genes from hundreds of individuals. This provides an extraordinary opportunity to learn phenotype relevant genomic patterns that can improve our understanding of molecular and cellular processes underlying a trait. The high dimensional nature of genomic data presents a range of computational and statistical challenges. This dissertation presents a compilation of projects that were driven by the motivation to efficiently capture gene regulatory patterns in the human transcriptome, while addressing statistical and computational challenges that accompany this data. We attempt to address two major difficulties in this domain: a) artifacts and noise in transcriptomic data, andb) limited statistical power. First, we present our work on investigating the effect of artifactual variation in gene expression data and its impact on trans-eQTL discovery. Here we performed an in-depth analysis of diverse pre-recorded covariates and latent confounders to understand their contribution to heterogeneity in gene expression measurements. Next, we discovered 673 trans-eQTLs across 16 human tissues using v6 data from the Genotype Tissue Expression (GTEx) project. Finally, we characterized two trait-associated trans-eQTLs; one in Skeletal Muscle and another in Thyroid. Second, we present a principal component based residualization method to correct gene expression measurements prior to reconstruction of co-expression networks. -

Supplemental Figures
A B Previously-induced Doxycycline-naïve survival (%) survival (%) Recurrence-free Recurrence-free Days following dox withdrawal Age C D Primary Recurrent ** Par-4 mRNA H2B-mCherry DAPI Primary Recurrent Supplemental Figure 1: Recurrent tumors are derived from primary tumors. A. Kaplan-Meier survival plot showing recurrent tumor-free survival in mice previously induced with doxycycline (n=30) or tumor-free survival in doxycycline-naïve mice (n=10). B. Kaplan-Meier survival plot showing recurrence-free survival following doxycycline withdrawal in a cohort of recipient mice with orthotopic tumors (n=5). C. Representative images (40x magnification) of primary and recurrent orthotopic tumors following injection of H2B-mCherry labeled primary tumor cell line #1 into recipient mice. D. qRT-PCR analysis of Par-4 transcripts from primary (n=5) and recurrent (n=5) orthotopic tumors. Significance determined by Student’s t-test. Error bars denote mean ± SEM. **p<0.01. A B 1 TWIST1 TWIST2 0.5 SNAI2 0 VIM -0.5 ZEB1 ZEB2 -1 SNAI1 PAWR Positively correlated Negatively correlated with Par-4 with Par-4 CDH1 CLDN7 CLDN4 CLDN3 KRT18 NES = -2.07335 q-value = 0.001129 KRT8 TWIST1 TWIST2 SNAI2 VIM ZEB1 ZEB2 SNAI1 PAWR CDH1 CLDN7 CLDN4 CLDN3 KRT18 KRT8 Marcotte, et al. C 1 SNAI1 0.5 TWIST1 TWIST2 0 VIM -0.5 SNAI2 -1 ZEB1 ZEB2 PAWR CDH1 KRT18 KRT8 CLDN7 CLDN3 CLDN4 SNAI1 TWIST1 TWIST2 VIM SNAI2 ZEB1 ZEB2 PAWR CDH1 KRT18 KRT8 CLDN7 CLDN3 CLDN4 TCGA, Cell 2015 Supplemental Figure 2: Par-4 expression is negatively correlated with EMT in human breast cancer. A. -

By Submitted in Partial Satisfaction of the Requirements for Degree of in In
Developments of Two Imaging based Technologies for Cell Biology Researches by Xiaowei Yan DISSERTATION Submitted in partial satisfaction of the requirements for degree of DOCTOR OF PHILOSOPHY in Biochemistry and Molecular Biology in the GRADUATE DIVISION of the UNIVERSITY OF CALIFORNIA, SAN FRANCISCO Approved: ______________________________________________________________________________Ronald Vale Chair ______________________________________________________________________________Jonathan Weissman ______________________________________________________________________________Orion Weiner ______________________________________________________________________________ ______________________________________________________________________________ Committee Members Copyright 2021 By Xiaowei Yan ii DEDICATION Everything happens for the best. To my family, who supported me with all their love. iii ACKNOWLEDGEMENTS The greatest joy of my PhD has been joining UCSF, working and learning with such a fantastic group of scientists. I am extremely grateful for all the support and mentorship I received and would like to thank: My mentor, Ron Vale, who is such a great and generous person. Thank you for showing me that science is so much fun and thank you for always giving me the freedom in pursuing my interest. I am grateful for all the guidance from you and thank you for always supporting me whenever I needed. You are a person full of wisdom, and I have been learning so much from you and your attitude to science, science community and even life will continue inspire me. Thank you for being my mentor and thank you for being such a great mentor. Everyone else in Vale lab, past and present, for making our lab a sweet home. I would like to give my special thank to Marvin (Marvin Tanenbaum) and Nico (Nico Stuurman), two other mentors for me in the lab. I would like to thank them for helping me adapt to our lab, for all the valuable advice and for all the happiness during the time that we work together. -
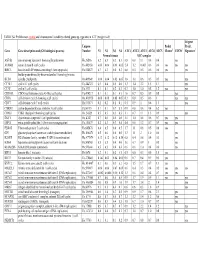
TABLE S4: Proliferation-Related and Chromosomal Instability-Related
TABLE S4: Proliferation-related and chromosomal instability-related genes up-regulated in ATC (weight <4.0) 44-gene Unigene Prolif. Prolif. Gene Gene description and (GO biological process) Number N1 N2 N3 N4 ATC1 ATC2 ATC3 ATC4 ATC5 Cluster1 CIN702 Signature3 Normal tissues ATC samples ASF1B anti-silencing function 1 homolog B (unknown) Hs.26516 0.2 0.3 0.3 0.3 0.8 0.8 1.1 0.8 0.4 yes AURKB aurora kinase B (cell cycle) Hs.442658 0.06 0.06 0.08 0.05 2.4 1.2 0.003 0.6 0.6 yes yes yes BIRC5 baculoviral IAP repeat-containing 5 (anti apoptosis) Hs.514527 0.7 0.3 0.4 0.3 0.8 0.5 0.5 0.6 0.6 yes yes budding uninhibited by benzimidazoles 1 homolog (mitotic BUB1 spindle checkpoint) Hs.469649 0.06 0.04 0.02 0.05 1.8 1.0 0.6 0.5 0.5 yes yes CCNE1 cyclin E1 (cell cycle) Hs.244723 0.5 0.4 0.5 0.6 1.3 1.4 2.2 1.5 1.1 yes CCNF cyclin F (cell cycle) Hs.1973 0.1 0.1 0.2 0.3 0.7 1.0 1.0 0.8 1.2 yes yes CDC45L CDC45 cell division cycle 45-like (cell cycle) Hs.474217 0.1 0.1 0.1 0.3 1.6 0.7 0.5 0.9 0.8 yes CDC6 cell division cycle 6 homolog (cell cycle) Hs. 405958 0.08 0.08 0.08 0.07 0.3 0.8 0.5 0.6 1 yes yes CDC7 cell division cycle 7 (cell cycle) Hs.533573 0.2 0.2 0.2 0.1 0.5 0.9 1 0.6 1.3 yes CDKN3 cyclin-dependent kinase inhibitor 3 (cell cycle) Hs.84113 0.1 0.1 0.7 0.1 0.9 0.8 0.6 0.4 6.2 yes CHEK1 CHK1 checkpoint homolog (cell cycle) Hs. -

1 AGING Supplementary Table 2
SUPPLEMENTARY TABLES Supplementary Table 1. Details of the eight domain chains of KIAA0101. Serial IDENTITY MAX IN COMP- INTERFACE ID POSITION RESOLUTION EXPERIMENT TYPE number START STOP SCORE IDENTITY LEX WITH CAVITY A 4D2G_D 52 - 69 52 69 100 100 2.65 Å PCNA X-RAY DIFFRACTION √ B 4D2G_E 52 - 69 52 69 100 100 2.65 Å PCNA X-RAY DIFFRACTION √ C 6EHT_D 52 - 71 52 71 100 100 3.2Å PCNA X-RAY DIFFRACTION √ D 6EHT_E 52 - 71 52 71 100 100 3.2Å PCNA X-RAY DIFFRACTION √ E 6GWS_D 41-72 41 72 100 100 3.2Å PCNA X-RAY DIFFRACTION √ F 6GWS_E 41-72 41 72 100 100 2.9Å PCNA X-RAY DIFFRACTION √ G 6GWS_F 41-72 41 72 100 100 2.9Å PCNA X-RAY DIFFRACTION √ H 6IIW_B 2-11 2 11 100 100 1.699Å UHRF1 X-RAY DIFFRACTION √ www.aging-us.com 1 AGING Supplementary Table 2. Significantly enriched gene ontology (GO) annotations (cellular components) of KIAA0101 in lung adenocarcinoma (LinkedOmics). Leading Description FDR Leading Edge Gene EdgeNum RAD51, SPC25, CCNB1, BIRC5, NCAPG, ZWINT, MAD2L1, SKA3, NUF2, BUB1B, CENPA, SKA1, AURKB, NEK2, CENPW, HJURP, NDC80, CDCA5, NCAPH, BUB1, ZWILCH, CENPK, KIF2C, AURKA, CENPN, TOP2A, CENPM, PLK1, ERCC6L, CDT1, CHEK1, SPAG5, CENPH, condensed 66 0 SPC24, NUP37, BLM, CENPE, BUB3, CDK2, FANCD2, CENPO, CENPF, BRCA1, DSN1, chromosome MKI67, NCAPG2, H2AFX, HMGB2, SUV39H1, CBX3, TUBG1, KNTC1, PPP1CC, SMC2, BANF1, NCAPD2, SKA2, NUP107, BRCA2, NUP85, ITGB3BP, SYCE2, TOPBP1, DMC1, SMC4, INCENP. RAD51, OIP5, CDK1, SPC25, CCNB1, BIRC5, NCAPG, ZWINT, MAD2L1, SKA3, NUF2, BUB1B, CENPA, SKA1, AURKB, NEK2, ESCO2, CENPW, HJURP, TTK, NDC80, CDCA5, BUB1, ZWILCH, CENPK, KIF2C, AURKA, DSCC1, CENPN, CDCA8, CENPM, PLK1, MCM6, ERCC6L, CDT1, HELLS, CHEK1, SPAG5, CENPH, PCNA, SPC24, CENPI, NUP37, FEN1, chromosomal 94 0 CENPL, BLM, KIF18A, CENPE, MCM4, BUB3, SUV39H2, MCM2, CDK2, PIF1, DNA2, region CENPO, CENPF, CHEK2, DSN1, H2AFX, MCM7, SUV39H1, MTBP, CBX3, RECQL4, KNTC1, PPP1CC, CENPP, CENPQ, PTGES3, NCAPD2, DYNLL1, SKA2, HAT1, NUP107, MCM5, MCM3, MSH2, BRCA2, NUP85, SSB, ITGB3BP, DMC1, INCENP, THOC3, XPO1, APEX1, XRCC5, KIF22, DCLRE1A, SEH1L, XRCC3, NSMCE2, RAD21. -

Emi1 (FBXO5) (Center) Rabbit Polyclonal Antibody Product Data
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for AP51634PU-N Emi1 (FBXO5) (Center) Rabbit Polyclonal Antibody Product data: Product Type: Primary Antibodies Applications: WB Recommended Dilution: ELISA: 1/1000. Western Blot: 1/100-1/500. Reactivity: Human Host: Rabbit Isotype: Ig Clonality: Polyclonal Immunogen: KLH conjugated synthetic peptide between 191-221 amino acids from the Central region of Human FBXO5. Specificity: This antibody recognizes Human FBXO5 (Center). Formulation: PBS containing 0.09% (W/V) Sodium Azide as preservative State: Aff - Purified State: Liquid purified Ig fraction Concentration: lot specific Purification: Affinity Chromatography on Protein A Conjugation: Unconjugated Storage: Store undiluted at 2-8°C for one month or (in aliquots) at -20°C for longer. Avoid repeated freezing and thawing. Stability: Shelf life: one year from despatch. Gene Name: Homo sapiens F-box protein 5 (FBXO5), transcript variant 2 Database Link: Entrez Gene 26271 Human Q9UKT4 This product is to be used for laboratory only. Not for diagnostic or therapeutic use. View online » ©2021 OriGene Technologies, Inc., 9620 Medical Center Drive, Ste 200, Rockville, MD 20850, US 1 / 2 Emi1 (FBXO5) (Center) Rabbit Polyclonal Antibody – AP51634PU-N Background: This gene encodes a member of the F-box protein family which is characterized by an approximately 40 amino acid motif, the F-box. The F-box proteins constitute one of the four subunits of the ubiquitin protein ligase complex called SCFs (SKP1-cullin-F-box), which function in phosphorylation-dependent ubiquitination. -

Supplementary Materials
Supplementary materials Supplementary Table S1: MGNC compound library Ingredien Molecule Caco- Mol ID MW AlogP OB (%) BBB DL FASA- HL t Name Name 2 shengdi MOL012254 campesterol 400.8 7.63 37.58 1.34 0.98 0.7 0.21 20.2 shengdi MOL000519 coniferin 314.4 3.16 31.11 0.42 -0.2 0.3 0.27 74.6 beta- shengdi MOL000359 414.8 8.08 36.91 1.32 0.99 0.8 0.23 20.2 sitosterol pachymic shengdi MOL000289 528.9 6.54 33.63 0.1 -0.6 0.8 0 9.27 acid Poricoic acid shengdi MOL000291 484.7 5.64 30.52 -0.08 -0.9 0.8 0 8.67 B Chrysanthem shengdi MOL004492 585 8.24 38.72 0.51 -1 0.6 0.3 17.5 axanthin 20- shengdi MOL011455 Hexadecano 418.6 1.91 32.7 -0.24 -0.4 0.7 0.29 104 ylingenol huanglian MOL001454 berberine 336.4 3.45 36.86 1.24 0.57 0.8 0.19 6.57 huanglian MOL013352 Obacunone 454.6 2.68 43.29 0.01 -0.4 0.8 0.31 -13 huanglian MOL002894 berberrubine 322.4 3.2 35.74 1.07 0.17 0.7 0.24 6.46 huanglian MOL002897 epiberberine 336.4 3.45 43.09 1.17 0.4 0.8 0.19 6.1 huanglian MOL002903 (R)-Canadine 339.4 3.4 55.37 1.04 0.57 0.8 0.2 6.41 huanglian MOL002904 Berlambine 351.4 2.49 36.68 0.97 0.17 0.8 0.28 7.33 Corchorosid huanglian MOL002907 404.6 1.34 105 -0.91 -1.3 0.8 0.29 6.68 e A_qt Magnogrand huanglian MOL000622 266.4 1.18 63.71 0.02 -0.2 0.2 0.3 3.17 iolide huanglian MOL000762 Palmidin A 510.5 4.52 35.36 -0.38 -1.5 0.7 0.39 33.2 huanglian MOL000785 palmatine 352.4 3.65 64.6 1.33 0.37 0.7 0.13 2.25 huanglian MOL000098 quercetin 302.3 1.5 46.43 0.05 -0.8 0.3 0.38 14.4 huanglian MOL001458 coptisine 320.3 3.25 30.67 1.21 0.32 0.9 0.26 9.33 huanglian MOL002668 Worenine -

Human Lectins, Their Carbohydrate Affinities and Where to Find Them
biomolecules Review Human Lectins, Their Carbohydrate Affinities and Where to Review HumanFind Them Lectins, Their Carbohydrate Affinities and Where to FindCláudia ThemD. Raposo 1,*, André B. Canelas 2 and M. Teresa Barros 1 1, 2 1 Cláudia D. Raposo * , Andr1 é LAQVB. Canelas‐Requimte,and Department M. Teresa of Chemistry, Barros NOVA School of Science and Technology, Universidade NOVA de Lisboa, 2829‐516 Caparica, Portugal; [email protected] 12 GlanbiaLAQV-Requimte,‐AgriChemWhey, Department Lisheen of Chemistry, Mine, Killoran, NOVA Moyne, School E41 of ScienceR622 Co. and Tipperary, Technology, Ireland; canelas‐ [email protected] NOVA de Lisboa, 2829-516 Caparica, Portugal; [email protected] 2* Correspondence:Glanbia-AgriChemWhey, [email protected]; Lisheen Mine, Tel.: Killoran, +351‐212948550 Moyne, E41 R622 Tipperary, Ireland; [email protected] * Correspondence: [email protected]; Tel.: +351-212948550 Abstract: Lectins are a class of proteins responsible for several biological roles such as cell‐cell in‐ Abstract:teractions,Lectins signaling are pathways, a class of and proteins several responsible innate immune for several responses biological against roles pathogens. such as Since cell-cell lec‐ interactions,tins are able signalingto bind to pathways, carbohydrates, and several they can innate be a immuneviable target responses for targeted against drug pathogens. delivery Since sys‐ lectinstems. In are fact, able several to bind lectins to carbohydrates, were approved they by canFood be and a viable Drug targetAdministration for targeted for drugthat purpose. delivery systems.Information In fact, about several specific lectins carbohydrate were approved recognition by Food by andlectin Drug receptors Administration was gathered for that herein, purpose. plus Informationthe specific organs about specific where those carbohydrate lectins can recognition be found by within lectin the receptors human was body. -

Differential Gene Expression in the Skin of Xiphophorus
DIFFERENTIAL GENE EXPRESSION IN THE SKIN OF XIPHOPHORUS MACULATUS JP 163 B IN RESPONSE TO FULL SPECTRUM (10,000 K) FLUORESCENT LIGHT by Kaela Caballero, B.S. A thesis submitted to the Graduate Council of Texas State University in partial fulfillment of the requirements for the degree of Master of Science with a Major in Biochemistry August 2015 Committee Members: Ronald Walter, Chair Rachell Booth Steven Whitten COPYRIGHT by Kaela L. Caballero 2015 FAIR USE AND AUTHOR’S PERMISSION STATEMENT Fair Use This work is protected by the Copyright Laws of the United States (Public Law 94-553, section 107). Consistent with fair use as defined in the Copyright Laws, brief quotations from this material are allowed with proper acknowledgment. Use of this material for financial gain without the author’s express written permission is not allowed. Duplication Permission As the copyright holder of this work I, Kaela L. Caballero, authorize duplication of this work, in whole or in part, for educational or scholarly purposes only. DEDICATION To my parents and family, who have always supported and encouraged me, my education and my coffee addiction. And to Max, my study buddy; where you have gone, there is no need for coffee fueled late nights. Rest in peace. ACKNOWLEDGEMENTS This work could not have been accomplished without the support and mentorship that I have received throughout my life from family, teachers, mentors and friends. It is impossible to name everyone here, but I would like to use a few lines to thank those that have helped me achieve my goals. Thank you to Dr. -

Expression of a Mirna Targeting Mutated SOD1 in Astrocytes Induces Motoneuron Plasticity and Improves Neuromuscular Function in ALS Mice
bioRxiv preprint doi: https://doi.org/10.1101/2021.01.08.425706; this version posted January 9, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Expression of a miRNA targeting mutated SOD1 in astrocytes induces motoneuron plasticity and improves neuromuscular function in ALS mice Rochat C.1, Bernard-Marissal N.1,6, Pradervand S.3, Perrin F.E.4, Raoul C.5, Aebischer P.1, Schneider B.L.1,2* 1 Brain Mind Institute, Ecole Polytechnique Fédérale de Lausanne (EPFL), 1015 Lausanne, Switzerland 2 Bertarelli Platform for Gene Therapy, Ecole Polytechnique Fédérale de Lausanne (EPFL), Geneva, Switzerland 3 Genomic Technologies Facility, University of Lausanne, Lausanne, Switzerland. 4 INSERM U1198, University of Montpellier, EPHE, Place Eugène Bataillon CC105, F-34095, Montpellier, France 5 The Neuroscience Institute of Montpellier, Inserm UMR1051, Univ Montpellier, Saint Eloi Hospital, Montpellier, France 6 Aix Marseille Univ, INSERM, MMG, Marseille, France *Correspondence should be addressed to B.L.S. ([email protected]) Short title: miRNA gene therapy targeting astrocytes in ALS Study was performed in Lausanne, Switzerland Current address of corresponding author: Bernard Schneider EPFL SV PTECH PTBTG Ch. des Mines 9 CH-1202 Genève Switzerland Email: [email protected] Tel: +41 21 693 95 05 bioRxiv preprint doi: https://doi.org/10.1101/2021.01.08.425706; this version posted January 9, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. -

Loss of the Mammalian APC/C Activator FZR1 Shortens G1 and Lengthens S Phase but Has Little Effect on Exit from Mitosis
4208 Research Article Loss of the mammalian APC/C activator FZR1 shortens G1 and lengthens S phase but has little effect on exit from mitosis Reinhard Sigl1, Cornelia Wandke1, Veronika Rauch1, Jane Kirk2, Tim Hunt2 and Stephan Geley1,* 1Division of Molecular Pathophysiology, Biocenter, Innsbruck Medical University, Innsbruck, Austria 2Clare Hall Laboratories, Cancer Research UK, South Mimms, England, UK *Author for correspondence ([email protected]) Accepted 10 September 2009 Journal of Cell Science 122, 4208-4217 Published by The Company of Biologists 2009 doi:10.1242/jcs.054197 Summary The anaphase-promoting complex/cyclosome (APC/C) is knockdown of p53 in U2OS cells with inducible FZR1 siRNA essential for progression through mitosis. At anaphase onset, also failed to restore their proliferative capacity. Thus, the the APC/C requires the activator protein CDC20 to target proliferation defects are a direct consequence of the genetic securin and cyclin B1 for proteasome-dependent degradation, damage inflicted by loss of FZR1 function and are largely but then depends on the CDC20-related protein FZR1 (also independent of p53. In summary, mammalian FZR1 is not known as CDH1) to remain active until the onset of the next S required for the completion of mitosis, but is an important phase. To investigate the role of FZR1 in mammalian cells, we regulator of G1 phase and is required for efficient DNA used RNAi in human cell lines and conditional gene targeting replication in human and mouse somatic cells. in mouse embryonic fibroblasts. In neither case was FZR1 required for exit from mitosis, but in cells lacking FZR1, the Supplementary material available online at G1 phase was shortened and the S phase was prolonged. -

FARE2021WINNERS Sorted by Institute
FARE2021WINNERS Sorted By Institute Swati Shah Postdoctoral Fellow CC Radiology/Imaging/PET and Neuroimaging Characterization of CNS involvement in Ebola-Infected Macaques using Magnetic Resonance Imaging, 18F-FDG PET and Immunohistology The Ebola (EBOV) virus outbreak in Western Africa resulted in residual neurologic abnormalities in survivors. Many case studies detected EBOV in the CSF, suggesting that the neurologic sequelae in survivors is related to viral presence. In the periphery, EBOV infects endothelial cells and triggers a “cytokine stormâ€. However, it is unclear whether a similar process occurs in the brain, with secondary neuroinflammation, neuronal loss and blood-brain barrier (BBB) compromise, eventually leading to lasting neurological damage. We have used in vivo imaging and post-necropsy immunostaining to elucidate the CNS pathophysiology in Rhesus macaques infected with EBOV (Makona). Whole brain MRI with T1 relaxometry (pre- and post-contrast) and FDG-PET were performed to monitor the progression of disease in two cohorts of EBOV infected macaques from baseline to terminal endpoint (day 5-6). Post-necropsy, multiplex fluorescence immunohistochemical (MF-IHC) staining for various cellular markers in the thalamus and brainstem was performed. Serial blood and CSF samples were collected to assess disease progression. The linear mixed effect model was used for statistical analysis. Post-infection, we first detected EBOV in the serum (day 3) and CSF (day 4) with dramatic increases until euthanasia. The standard uptake values of FDG-PET relative to whole brain uptake (SUVr) in the midbrain, pons, and thalamus increased significantly over time (p<0.01) and positively correlated with blood viremia (p≤0.01).