A Systems-Wide Screen Identifies Substrates of the SCF Ubiquitin Ligase
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Prevalence and Functional Analysis of Sequence Variants in the ATR Checkpoint Mediator Claspin
Published OnlineFirst September 8, 2009; DOI: 10.1158/1541-7786.MCR-09-0033 Prevalence and Functional Analysis of Sequence Variants in the ATR Checkpoint Mediator Claspin Jianmin Zhang,1 Young-Han Song,1 Brian W. Brannigan,1 Doke C.R. Wahrer,1 Taryn A. Schiripo,1 Patricia L. Harris,1 Sara M. Haserlat,1 Lindsey E. Ulkus,1 Kristen M. Shannon,1 Judy E. Garber,2 Matthew L. Freedman,3 Brian E. Henderson,4 Lee Zou,1 Dennis C. Sgroi,1 Daniel A. Haber,1 and Daphne W. Bell1 1Massachusetts General Hospital Cancer Center and Harvard Medical School, Charlestown, Massachusetts; 2Dana-Farber Cancer Institute and Harvard Medical School, Boston, Massachusetts; 3Department of Molecular Biology, Massachusetts General Hospital, Department of Genetics, Harvard Medical School, and Broad Institute for Biomedical Research, Boston, Massachusetts; and 4Department of Preventive Medicine, University of Southern California Keck School of Medicine, Los Angeles, California Abstract mutation, was defective in its ability to mediate CHK1 Mutational inactivation of genes controlling the phosphorylation followingDNA damageand was unable to DNA-damage response contributes to cancer susceptibility rescue sensitivity to replicative stress in CLSPN-depleted within families and within the general population as well as cells. Taken together, these observations raise the to sporadic tumorigenesis. Claspin (CLSPN) encodes a possibility that CLSPN may encode a component of the recently recognized mediator protein essential for the ATR DNA-damage response pathway that is targeted by and CHK1-dependent checkpoint elicited by replicative mutations in human cancers, suggesting the need for larger stress or the presence of ssDNA. Here, we describe a study population-based studies to investigate whether CLSPN to determine whether mutational disruption of CLSPN variants contribute to cancer susceptibility. -

PARSANA-DISSERTATION-2020.Pdf
DECIPHERING TRANSCRIPTIONAL PATTERNS OF GENE REGULATION: A COMPUTATIONAL APPROACH by Princy Parsana A dissertation submitted to The Johns Hopkins University in conformity with the requirements for the degree of Doctor of Philosophy Baltimore, Maryland July, 2020 © 2020 Princy Parsana All rights reserved Abstract With rapid advancements in sequencing technology, we now have the ability to sequence the entire human genome, and to quantify expression of tens of thousands of genes from hundreds of individuals. This provides an extraordinary opportunity to learn phenotype relevant genomic patterns that can improve our understanding of molecular and cellular processes underlying a trait. The high dimensional nature of genomic data presents a range of computational and statistical challenges. This dissertation presents a compilation of projects that were driven by the motivation to efficiently capture gene regulatory patterns in the human transcriptome, while addressing statistical and computational challenges that accompany this data. We attempt to address two major difficulties in this domain: a) artifacts and noise in transcriptomic data, andb) limited statistical power. First, we present our work on investigating the effect of artifactual variation in gene expression data and its impact on trans-eQTL discovery. Here we performed an in-depth analysis of diverse pre-recorded covariates and latent confounders to understand their contribution to heterogeneity in gene expression measurements. Next, we discovered 673 trans-eQTLs across 16 human tissues using v6 data from the Genotype Tissue Expression (GTEx) project. Finally, we characterized two trait-associated trans-eQTLs; one in Skeletal Muscle and another in Thyroid. Second, we present a principal component based residualization method to correct gene expression measurements prior to reconstruction of co-expression networks. -

Assembly and Maintenance of Sarcomere Thin Filaments and Associated Diseases
International Journal of Molecular Sciences Review Assembly and Maintenance of Sarcomere Thin Filaments and Associated Diseases Kendal Prill and John F. Dawson * Centre for Cardiovascular Investigations, Department of Molecular and Cellular Biology, University of Guelph, Guelph, ON N1G 2W1, Canada; [email protected] * Correspondence: [email protected] Received: 17 December 2019; Accepted: 12 January 2020; Published: 15 January 2020 Abstract: Sarcomere assembly and maintenance are essential physiological processes required for cardiac and skeletal muscle function and organism mobility. Over decades of research, components of the sarcomere and factors involved in the formation and maintenance of this contractile unit have been identified. Although we have a general understanding of sarcomere assembly and maintenance, much less is known about the development of the thin filaments and associated factors within the sarcomere. In the last decade, advancements in medical intervention and genome sequencing have uncovered patients with novel mutations in sarcomere thin filaments. Pairing this sequencing with reverse genetics and the ability to generate patient avatars in model organisms has begun to deepen our understanding of sarcomere thin filament development. In this review, we provide a summary of recent findings regarding sarcomere assembly, maintenance, and disease with respect to thin filaments, building on the previous knowledge in the field. We highlight debated and unknown areas within these processes to clearly define open research questions. Keywords: sarcomere assembly; sarcomere maintenance; sarcomere thin filaments; thin filament assembly; thin filament maintenance; thin filament turnover; actin turnover; chaperone; sarcomere; myopathy 1. Introduction Striated muscle requires the coordination of hundreds of proteins not only for cellular function but also for assembly of the contractile sarcomere units within the myofibril. -

By Submitted in Partial Satisfaction of the Requirements for Degree of in In
Developments of Two Imaging based Technologies for Cell Biology Researches by Xiaowei Yan DISSERTATION Submitted in partial satisfaction of the requirements for degree of DOCTOR OF PHILOSOPHY in Biochemistry and Molecular Biology in the GRADUATE DIVISION of the UNIVERSITY OF CALIFORNIA, SAN FRANCISCO Approved: ______________________________________________________________________________Ronald Vale Chair ______________________________________________________________________________Jonathan Weissman ______________________________________________________________________________Orion Weiner ______________________________________________________________________________ ______________________________________________________________________________ Committee Members Copyright 2021 By Xiaowei Yan ii DEDICATION Everything happens for the best. To my family, who supported me with all their love. iii ACKNOWLEDGEMENTS The greatest joy of my PhD has been joining UCSF, working and learning with such a fantastic group of scientists. I am extremely grateful for all the support and mentorship I received and would like to thank: My mentor, Ron Vale, who is such a great and generous person. Thank you for showing me that science is so much fun and thank you for always giving me the freedom in pursuing my interest. I am grateful for all the guidance from you and thank you for always supporting me whenever I needed. You are a person full of wisdom, and I have been learning so much from you and your attitude to science, science community and even life will continue inspire me. Thank you for being my mentor and thank you for being such a great mentor. Everyone else in Vale lab, past and present, for making our lab a sweet home. I would like to give my special thank to Marvin (Marvin Tanenbaum) and Nico (Nico Stuurman), two other mentors for me in the lab. I would like to thank them for helping me adapt to our lab, for all the valuable advice and for all the happiness during the time that we work together. -

The Malignant Phenotype in Breast Cancer Is Driven by Eif4a1-Mediated Changes in the Translational Landscape
Citation: Cell Death and Disease (2015) 6, e1603; doi:10.1038/cddis.2014.542 OPEN & 2015 Macmillan Publishers Limited All rights reserved 2041-4889/15 www.nature.com/cddis The malignant phenotype in breast cancer is driven by eIF4A1-mediated changes in the translational landscape A Modelska1, E Turro1,2, R Russell1, J Beaton1, T Sbarrato3, K Spriggs4, J Miller1, S Gräf1,2,5, E Provenzano6,7, F Blows8, P Pharoah6,8, C Caldas1,6 and J Le Quesne*,1,3 Human mRNA DeXD/H-box helicases are ubiquitous molecular motors that are required for the majority of cellular processes that involve RNA metabolism. One of the most abundant is eIF4A, which is required during the initiation phase of protein synthesis to unwind regions of highly structured mRNA that would otherwise impede the scanning ribosome. Dysregulation of protein synthesis is associated with tumorigenesis, but little is known about the detailed relationships between RNA helicase function and the malignant phenotype in solid malignancies. Therefore, immunohistochemical analysis was performed on over 3000 breast tumors to investigate the relationship among expression of eIF4A1, the helicase-modulating proteins eIF4B, eIF4E and PDCD4, and clinical outcome. We found eIF4A1, eIF4B and eIF4E to be independent predictors of poor outcome in ER-negative disease, while in contrast, the eIF4A1 inhibitor PDCD4 was related to improved outcome in ER-positive breast cancer. Consistent with these data, modulation of eIF4A1, eIF4B and PCDC4 expression in cultured MCF7 cells all restricted breast cancer cell growth and cycling. The eIF4A1-dependent translatome of MCF7 cells was defined by polysome profiling, and was shown to be highly enriched for several classes of oncogenic genes, including G-protein constituents, cyclins and protein kinases, and for mRNAs with G/C-rich 5′UTRs with potential to form G-quadruplexes and with 3′UTRs containing microRNA target sites. -

ATR Pathway Inhibition Is Synthetically Lethal in Cancer Cells with ERCC1 Deficiency
Published OnlineFirst March 24, 2014; DOI: 10.1158/0008-5472.CAN-13-3229 Cancer Therapeutics, Targets, and Chemical Biology Research ATR Pathway Inhibition Is Synthetically Lethal in Cancer Cells with ERCC1 Deficiency Kareem N. Mohni, Gina M. Kavanaugh, and David Cortez Abstract The DNA damage response kinase ATR and its effector kinase CHEK1 are required for cancer cells to survive oncogene-induced replication stress. ATR inhibitors exhibit synthetic lethal interactions, with deficiencies in the DNA damage response enzymes ATM and XRCC1 and with overexpression of the cell cycle kinase cyclin E. Here, we report a systematic screen to identify synthetic lethal interactions with ATR pathway–targeted drugs, rationalized by their predicted therapeutic utility in the oncology clinic. We found that reduced function in the ATR pathway itself provided the strongest synthetic lethal interaction. In addition, we found that loss of the structure-specific endonuclease ERCC1-XPF (ERCC4) is synthetic lethal with ATR pathway inhibitors. ERCC1- deficient cells exhibited elevated levels of DNA damage, which was increased further by ATR inhibition. When treated with ATR or CHEK1 inhibitors, ERCC1-deficient cells were arrested in S-phase and failed to complete cell-cycle transit even after drug removal. Notably, triple-negative breast cancer cells and non–small cell lung cancer cells depleted of ERCC1 exhibited increased sensitivity to ATR pathway–targeted drugs. Overall, we concluded that ATR pathway–targeted drugs may offer particular utility in cancers with reduced ATR pathway function or reduced levels of ERCC4 activity. Cancer Res; 74(10); 1–11. Ó2014 AACR. Introduction repair pathway such as homologous recombination or post- – Replicating DNA is sensitive to a wide array of endogenous replicative repair to remove the PARP DNA complexes (7). -

4-6 Weeks Old Female C57BL/6 Mice Obtained from Jackson Labs Were Used for Cell Isolation
Methods Mice: 4-6 weeks old female C57BL/6 mice obtained from Jackson labs were used for cell isolation. Female Foxp3-IRES-GFP reporter mice (1), backcrossed to B6/C57 background for 10 generations, were used for the isolation of naïve CD4 and naïve CD8 cells for the RNAseq experiments. The mice were housed in pathogen-free animal facility in the La Jolla Institute for Allergy and Immunology and were used according to protocols approved by the Institutional Animal Care and use Committee. Preparation of cells: Subsets of thymocytes were isolated by cell sorting as previously described (2), after cell surface staining using CD4 (GK1.5), CD8 (53-6.7), CD3ε (145- 2C11), CD24 (M1/69) (all from Biolegend). DP cells: CD4+CD8 int/hi; CD4 SP cells: CD4CD3 hi, CD24 int/lo; CD8 SP cells: CD8 int/hi CD4 CD3 hi, CD24 int/lo (Fig S2). Peripheral subsets were isolated after pooling spleen and lymph nodes. T cells were enriched by negative isolation using Dynabeads (Dynabeads untouched mouse T cells, 11413D, Invitrogen). After surface staining for CD4 (GK1.5), CD8 (53-6.7), CD62L (MEL-14), CD25 (PC61) and CD44 (IM7), naïve CD4+CD62L hiCD25-CD44lo and naïve CD8+CD62L hiCD25-CD44lo were obtained by sorting (BD FACS Aria). Additionally, for the RNAseq experiments, CD4 and CD8 naïve cells were isolated by sorting T cells from the Foxp3- IRES-GFP mice: CD4+CD62LhiCD25–CD44lo GFP(FOXP3)– and CD8+CD62LhiCD25– CD44lo GFP(FOXP3)– (antibodies were from Biolegend). In some cases, naïve CD4 cells were cultured in vitro under Th1 or Th2 polarizing conditions (3, 4). -

Substrate Trapping Proteomics Reveals Targets of the Trcp2
Substrate Trapping Proteomics Reveals Targets of the TrCP2/FBXW11 Ubiquitin Ligase Tai Young Kim,a,b* Priscila F. Siesser,a,b Kent L. Rossman,b,c Dennis Goldfarb,a,d Kathryn Mackinnon,a,b Feng Yan,a,b XianHua Yi,e Michael J. MacCoss,e Randall T. Moon,f Channing J. Der,b,c Michael B. Majora,b,d Department of Cell Biology and Physiology,a Lineberger Comprehensive Cancer Center,b Department of Pharmacology,c and Department of Computer Science,d University of North Carolina at Chapel Hill, Chapel Hill, North Carolina, USA; Department of Genome Sciencese and Department of Pharmacology and HHMI,f University of Washington, Seattle, Washington, USA Defining the full complement of substrates for each ubiquitin ligase remains an important challenge. Improvements in mass spectrometry instrumentation and computation and in protein biochemistry methods have resulted in several new methods for ubiquitin ligase substrate identification. Here we used the parallel adapter capture (PAC) proteomics approach to study TrCP2/FBXW11, a substrate adaptor for the SKP1–CUL1–F-box (SCF) E3 ubiquitin ligase complex. The processivity of the ubiquitylation reaction necessitates transient physical interactions between FBXW11 and its substrates, thus making biochemi- cal purification of FBXW11-bound substrates difficult. Using the PAC-based approach, we inhibited the proteasome to “trap” ubiquitylated substrates on the SCFFBXW11 E3 complex. Comparative mass spectrometry analysis of immunopurified FBXW11 protein complexes before and after proteasome inhibition revealed 21 known and 23 putatively novel substrates. In focused studies, we found that SCFFBXW11 bound, polyubiquitylated, and destabilized RAPGEF2, a guanine nucleotide exchange factor that activates the small GTPase RAP1. -

Effects of Rapamycin on Social Interaction Deficits and Gene
Kotajima-Murakami et al. Molecular Brain (2019) 12:3 https://doi.org/10.1186/s13041-018-0423-2 RESEARCH Open Access Effects of rapamycin on social interaction deficits and gene expression in mice exposed to valproic acid in utero Hiroko Kotajima-Murakami1,2, Toshiyuki Kobayashi3, Hirofumi Kashii1,4, Atsushi Sato1,5, Yoko Hagino1, Miho Tanaka1,6, Yasumasa Nishito7, Yukio Takamatsu7, Shigeo Uchino1,2 and Kazutaka Ikeda1* Abstract The mammalian target of rapamycin (mTOR) signaling pathway plays a crucial role in cell metabolism, growth, and proliferation. The overactivation of mTOR has been implicated in the pathogenesis of syndromic autism spectrum disorder (ASD), such as tuberous sclerosis complex (TSC). Treatment with the mTOR inhibitor rapamycin improved social interaction deficits in mouse models of TSC. Prenatal exposure to valproic acid (VPA) increases the incidence of ASD. Rodent pups that are exposed to VPA in utero have been used as an animal model of ASD. Activation of the mTOR signaling pathway was recently observed in rodents that were exposed to VPA in utero, and rapamycin ameliorated social interaction deficits. The present study investigated the effect of rapamycin on social interaction deficits in both adolescence and adulthood, and gene expressions in mice that were exposed to VPA in utero. We subcutaneously injected 600 mg/kg VPA in pregnant mice on gestational day 12.5 and used the pups as a model of ASD. The pups were intraperitoneally injected with rapamycin or an equal volume of vehicle once daily for 2 consecutive days. The social interaction test was conducted in the offspring after the last rapamycin administration at 5–6 weeks of ages (adolescence) or 10–11 weeks of age (adulthood). -

1 Isolation and Characterization of Cul1-7, a Recessive Allele Of
Genetics: Published Articles Ahead of Print, published on February 25, 2009 as 10.1534/genetics.108.097675 Isolation and Characterization of cul1-7, a Recessive Allele of CULLIN1 that Disrupts SCF Function at the C-terminus of CUL1 in Arabidopsis thaliana Jonathan Gilkerson*,†, Jianhong Hu‡, Jessica Brown*, Alexander Jones* 1, Tai-ping Sun‡ and Judy Callis*,†,2 *Department of Molecular and Cellular Biology and †Plant Biology Graduate Group, University of California-Davis, Davis, CA 95616, ‡Department of Biology, Duke University, Durham, North Carolina 27708-1000 1 Current Address: Department of Plant and Microbial Biology, UC-Berkeley, Berkeley, CA 94720-3102 1 Running head: Characterization of cul1-7 Key words: Protein Degradation, Aux/IAA, RGA, SCF Ubiquitin Ligase, RBX1 2Corresponding Author: Judy Callis Department of Molecular and Cellular Biology University of California-Davis One Shields Avenue Davis, CA 95616 Phone: 530-752-1015 Fax: 530-752-3085 E-mail: [email protected] 2 ABSTRACT Many aspects of plant biology depend on the ubiquitin proteasome system for degradation of regulatory proteins. Ubiquitin E3 ligases confer substrate specificity in this pathway, and SCF-type ligases comprise a major class of E3s. SCF ligases have four subunits: SKP1, CUL1, RBX1, and an F-box protein for substrate recognition. The Aux/IAAs are a well- characterized family of SCF substrates in plants. Here, we report characterization of a mutant isolated from a genetic screen in Arabidopsis thaliana designed to identify plants defective in degradation of an Aux/IAA fusion protein, Aux/IAA1-luciferase (IAA1-LUC). This mutant exhibited four-fold slower IAA1-LUC degradation compared to the progenitor line, and seedlings displayed altered auxin responses. -
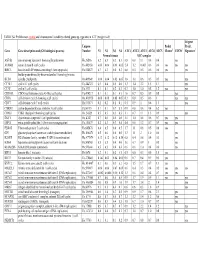
TABLE S4: Proliferation-Related and Chromosomal Instability-Related
TABLE S4: Proliferation-related and chromosomal instability-related genes up-regulated in ATC (weight <4.0) 44-gene Unigene Prolif. Prolif. Gene Gene description and (GO biological process) Number N1 N2 N3 N4 ATC1 ATC2 ATC3 ATC4 ATC5 Cluster1 CIN702 Signature3 Normal tissues ATC samples ASF1B anti-silencing function 1 homolog B (unknown) Hs.26516 0.2 0.3 0.3 0.3 0.8 0.8 1.1 0.8 0.4 yes AURKB aurora kinase B (cell cycle) Hs.442658 0.06 0.06 0.08 0.05 2.4 1.2 0.003 0.6 0.6 yes yes yes BIRC5 baculoviral IAP repeat-containing 5 (anti apoptosis) Hs.514527 0.7 0.3 0.4 0.3 0.8 0.5 0.5 0.6 0.6 yes yes budding uninhibited by benzimidazoles 1 homolog (mitotic BUB1 spindle checkpoint) Hs.469649 0.06 0.04 0.02 0.05 1.8 1.0 0.6 0.5 0.5 yes yes CCNE1 cyclin E1 (cell cycle) Hs.244723 0.5 0.4 0.5 0.6 1.3 1.4 2.2 1.5 1.1 yes CCNF cyclin F (cell cycle) Hs.1973 0.1 0.1 0.2 0.3 0.7 1.0 1.0 0.8 1.2 yes yes CDC45L CDC45 cell division cycle 45-like (cell cycle) Hs.474217 0.1 0.1 0.1 0.3 1.6 0.7 0.5 0.9 0.8 yes CDC6 cell division cycle 6 homolog (cell cycle) Hs. 405958 0.08 0.08 0.08 0.07 0.3 0.8 0.5 0.6 1 yes yes CDC7 cell division cycle 7 (cell cycle) Hs.533573 0.2 0.2 0.2 0.1 0.5 0.9 1 0.6 1.3 yes CDKN3 cyclin-dependent kinase inhibitor 3 (cell cycle) Hs.84113 0.1 0.1 0.7 0.1 0.9 0.8 0.6 0.4 6.2 yes CHEK1 CHK1 checkpoint homolog (cell cycle) Hs. -

Stockholm University
Stockholm University This is a published version of a paper published in Journal of Molecular Biology. Citation for the published paper: Björklund, Å., Light, S., Sagit, R., Elofsson, A. (2010) "Nebulin: A Study of Protein Repeat Evolution" Journal of Molecular Biology, 402(1): 38-51 URL: http://dx.doi.org/10.1016/j.jmb.2010.07.011 Access to the published version may require subscription. Permanent link to this version: http://urn.kb.se/resolve?urn=urn:nbn:se:su:diva-50177 http://su.diva-portal.org RGR YJMBI-62434; No. of pages: 14; 4C: 2, 5, 8, 9 doi:10.1016/j.jmb.2010.07.011 J. Mol. Biol. (2010) xx, xxx–xxx Available online at www.sciencedirect.com Nebulin: A Study of Protein Repeat Evolution Åsa K. Björklund, Sara Light†, Rauan Sagit† and Arne Elofsson⁎ Center for Biological Membrane Protein domain repeats are common in proteins that are central to the Research, Stockholm organization of a cell, in particular in eukaryotes. They are known to evolve Bioinformatics Center, through internal tandem duplications. However, the understanding of the Department of Biochemistry and underlying mechanisms is incomplete. To shed light on repeat expansion Biophysics, Stockholm mechanisms, we have studied the evolution of the muscle protein Nebulin, University, Stockholm, Sweden a protein that contains a large number of actin-binding nebulin domains. Nebulin proteins have evolved from an invertebrate precursor containing Received 30 March 2010; two nebulin domains. Repeat regions have expanded through duplications received in revised form of single domains, as well as duplications of a super repeat (SR) consisting 22 June 2010; of seven nebulins.