Expression of a Mirna Targeting Mutated SOD1 in Astrocytes Induces Motoneuron Plasticity and Improves Neuromuscular Function in ALS Mice
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

PARSANA-DISSERTATION-2020.Pdf
DECIPHERING TRANSCRIPTIONAL PATTERNS OF GENE REGULATION: A COMPUTATIONAL APPROACH by Princy Parsana A dissertation submitted to The Johns Hopkins University in conformity with the requirements for the degree of Doctor of Philosophy Baltimore, Maryland July, 2020 © 2020 Princy Parsana All rights reserved Abstract With rapid advancements in sequencing technology, we now have the ability to sequence the entire human genome, and to quantify expression of tens of thousands of genes from hundreds of individuals. This provides an extraordinary opportunity to learn phenotype relevant genomic patterns that can improve our understanding of molecular and cellular processes underlying a trait. The high dimensional nature of genomic data presents a range of computational and statistical challenges. This dissertation presents a compilation of projects that were driven by the motivation to efficiently capture gene regulatory patterns in the human transcriptome, while addressing statistical and computational challenges that accompany this data. We attempt to address two major difficulties in this domain: a) artifacts and noise in transcriptomic data, andb) limited statistical power. First, we present our work on investigating the effect of artifactual variation in gene expression data and its impact on trans-eQTL discovery. Here we performed an in-depth analysis of diverse pre-recorded covariates and latent confounders to understand their contribution to heterogeneity in gene expression measurements. Next, we discovered 673 trans-eQTLs across 16 human tissues using v6 data from the Genotype Tissue Expression (GTEx) project. Finally, we characterized two trait-associated trans-eQTLs; one in Skeletal Muscle and another in Thyroid. Second, we present a principal component based residualization method to correct gene expression measurements prior to reconstruction of co-expression networks. -

A New Genetic Method for Isolating Functionally Interacting Genes
Copyright 2000 by the Genetics Society of America A New Genetic Method for Isolating Functionally Interacting Genes: High plo1؉-Dependent Mutants and Their Suppressors De®ne Genes in Mitotic and Septation Pathways in Fission Yeast C. Fiona Cullen,*,² Karen M. May,* Iain M. Hagan,³ David M. Glover²,§ and Hiroyuki Ohkura*,² *Institute of Cell and Molecular Biology, The University of Edinburgh, Edinburgh EH9 3JR, United Kingdom, ²Department of Anatomy and Physiology, Medical Sciences Institute, The University of Dundee, Dundee DD1 4HN, United Kingdom, ³School of Biological Sciences, The University of Manchester, Manchester M13 9PT, United Kingdom and §Department of Genetics, University of Cambridge, Cambridge CB2 3EH, United Kingdom Manuscript received February 2, 2000 Accepted for publication April 10, 2000 ABSTRACT We describe a general genetic method to identify genes encoding proteins that functionally interact with and/or are good candidates for downstream targets of a particular gene product. The screen identi®es mutants whose growth depends on high levels of expression of that gene. We apply this to the plo1ϩ gene that encodes a ®ssion yeast homologue of the polo-like kinases. plo1ϩ regulates both spindle formation and septation. We have isolated 17 high plo1ϩ-dependent (pld) mutants that show defects in mitosis or septation. Three mutants show a mitotic arrest phenotype. Among the 14 pld mutants with septation defects, 12 mapped to known loci: cdc7, cdc15, cdc11 spg1, and sid2. One of the pld mutants, cdc7-PD1, was selected for suppressor analysis. As multicopy suppressors, we isolated four known genes involved in septation in ®ssion yeast: spg1ϩ, sce3ϩ, cdc8ϩ, and rho1ϩ, and two previously uncharacterized genes, mpd1ϩ and mpd2ϩ. -

By Submitted in Partial Satisfaction of the Requirements for Degree of in In
Developments of Two Imaging based Technologies for Cell Biology Researches by Xiaowei Yan DISSERTATION Submitted in partial satisfaction of the requirements for degree of DOCTOR OF PHILOSOPHY in Biochemistry and Molecular Biology in the GRADUATE DIVISION of the UNIVERSITY OF CALIFORNIA, SAN FRANCISCO Approved: ______________________________________________________________________________Ronald Vale Chair ______________________________________________________________________________Jonathan Weissman ______________________________________________________________________________Orion Weiner ______________________________________________________________________________ ______________________________________________________________________________ Committee Members Copyright 2021 By Xiaowei Yan ii DEDICATION Everything happens for the best. To my family, who supported me with all their love. iii ACKNOWLEDGEMENTS The greatest joy of my PhD has been joining UCSF, working and learning with such a fantastic group of scientists. I am extremely grateful for all the support and mentorship I received and would like to thank: My mentor, Ron Vale, who is such a great and generous person. Thank you for showing me that science is so much fun and thank you for always giving me the freedom in pursuing my interest. I am grateful for all the guidance from you and thank you for always supporting me whenever I needed. You are a person full of wisdom, and I have been learning so much from you and your attitude to science, science community and even life will continue inspire me. Thank you for being my mentor and thank you for being such a great mentor. Everyone else in Vale lab, past and present, for making our lab a sweet home. I would like to give my special thank to Marvin (Marvin Tanenbaum) and Nico (Nico Stuurman), two other mentors for me in the lab. I would like to thank them for helping me adapt to our lab, for all the valuable advice and for all the happiness during the time that we work together. -

HERV-K(HML7) Integrations in the Human Genome: Comprehensive Characterization and Comparative Analysis in Non-Human Primates
biology Article HERV-K(HML7) Integrations in the Human Genome: Comprehensive Characterization and Comparative Analysis in Non-Human Primates Nicole Grandi 1,* , Maria Paola Pisano 1 , Eleonora Pessiu 1, Sante Scognamiglio 1 and Enzo Tramontano 1,2 1 Laboratory of Molecular Virology, Department of Life and Environmental Sciences, University of Cagliari, 09042 Monserrato, Cagliari, Italy; [email protected] (M.P.P.); [email protected] (E.P.); [email protected] (S.S.); [email protected] (E.T.) 2 Istituto di Ricerca Genetica e Biomedica, Consiglio Nazionale delle Ricerche (CNR), 09042 Monserrato, Cagliari, Italy * Correspondence: [email protected] Simple Summary: The human genome is not human at all, but it includes a multitude of sequences inherited from ancient viral infections that affected primates’ germ line. These elements can be seen as the fossils of now-extinct retroviruses, and are called Human Endogenous Retroviruses (HERVs). View as “junk DNA” for a long time, HERVs constitute 4 times the amount of DNA needed to produce all cellular proteins, and growing evidence indicates their crucial role in primate brain evolution, placenta development, and innate immunity shaping. HERVs are also intensively studied for a pathological role, even if the incomplete knowledge about their exact number and genomic position has thus far prevented any causal association. Among possible relevant HERVs, the HERV-K Citation: Grandi, N.; Pisano, M.P.; supergroup is of particular interest, including some of the oldest (HML5) as well as youngest (HML2) Pessiu, E.; Scognamiglio, S.; integrations. Among HERV-Ks, the HML7 group still lack a detailed description, and the present Tramontano, E. -

Integrating Single-Step GWAS and Bipartite Networks Reconstruction Provides Novel Insights Into Yearling Weight and Carcass Traits in Hanwoo Beef Cattle
animals Article Integrating Single-Step GWAS and Bipartite Networks Reconstruction Provides Novel Insights into Yearling Weight and Carcass Traits in Hanwoo Beef Cattle Masoumeh Naserkheil 1 , Abolfazl Bahrami 1 , Deukhwan Lee 2,* and Hossein Mehrban 3 1 Department of Animal Science, University College of Agriculture and Natural Resources, University of Tehran, Karaj 77871-31587, Iran; [email protected] (M.N.); [email protected] (A.B.) 2 Department of Animal Life and Environment Sciences, Hankyong National University, Jungang-ro 327, Anseong-si, Gyeonggi-do 17579, Korea 3 Department of Animal Science, Shahrekord University, Shahrekord 88186-34141, Iran; [email protected] * Correspondence: [email protected]; Tel.: +82-31-670-5091 Received: 25 August 2020; Accepted: 6 October 2020; Published: 9 October 2020 Simple Summary: Hanwoo is an indigenous cattle breed in Korea and popular for meat production owing to its rapid growth and high-quality meat. Its yearling weight and carcass traits (backfat thickness, carcass weight, eye muscle area, and marbling score) are economically important for the selection of young and proven bulls. In recent decades, the advent of high throughput genotyping technologies has made it possible to perform genome-wide association studies (GWAS) for the detection of genomic regions associated with traits of economic interest in different species. In this study, we conducted a weighted single-step genome-wide association study which combines all genotypes, phenotypes and pedigree data in one step (ssGBLUP). It allows for the use of all SNPs simultaneously along with all phenotypes from genotyped and ungenotyped animals. Our results revealed 33 relevant genomic regions related to the traits of interest. -
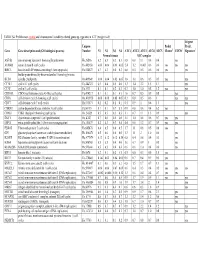
TABLE S4: Proliferation-Related and Chromosomal Instability-Related
TABLE S4: Proliferation-related and chromosomal instability-related genes up-regulated in ATC (weight <4.0) 44-gene Unigene Prolif. Prolif. Gene Gene description and (GO biological process) Number N1 N2 N3 N4 ATC1 ATC2 ATC3 ATC4 ATC5 Cluster1 CIN702 Signature3 Normal tissues ATC samples ASF1B anti-silencing function 1 homolog B (unknown) Hs.26516 0.2 0.3 0.3 0.3 0.8 0.8 1.1 0.8 0.4 yes AURKB aurora kinase B (cell cycle) Hs.442658 0.06 0.06 0.08 0.05 2.4 1.2 0.003 0.6 0.6 yes yes yes BIRC5 baculoviral IAP repeat-containing 5 (anti apoptosis) Hs.514527 0.7 0.3 0.4 0.3 0.8 0.5 0.5 0.6 0.6 yes yes budding uninhibited by benzimidazoles 1 homolog (mitotic BUB1 spindle checkpoint) Hs.469649 0.06 0.04 0.02 0.05 1.8 1.0 0.6 0.5 0.5 yes yes CCNE1 cyclin E1 (cell cycle) Hs.244723 0.5 0.4 0.5 0.6 1.3 1.4 2.2 1.5 1.1 yes CCNF cyclin F (cell cycle) Hs.1973 0.1 0.1 0.2 0.3 0.7 1.0 1.0 0.8 1.2 yes yes CDC45L CDC45 cell division cycle 45-like (cell cycle) Hs.474217 0.1 0.1 0.1 0.3 1.6 0.7 0.5 0.9 0.8 yes CDC6 cell division cycle 6 homolog (cell cycle) Hs. 405958 0.08 0.08 0.08 0.07 0.3 0.8 0.5 0.6 1 yes yes CDC7 cell division cycle 7 (cell cycle) Hs.533573 0.2 0.2 0.2 0.1 0.5 0.9 1 0.6 1.3 yes CDKN3 cyclin-dependent kinase inhibitor 3 (cell cycle) Hs.84113 0.1 0.1 0.7 0.1 0.9 0.8 0.6 0.4 6.2 yes CHEK1 CHK1 checkpoint homolog (cell cycle) Hs. -

Multiple Hits for the Association of Uterine Fibroids on Human Chromosome 1Q43
Multiple Hits for the Association of Uterine Fibroids on Human Chromosome 1q43 Brahim Aissani1*, Howard Wiener1, Kui Zhang2 1 Department of Epidemiology, University of Alabama at Birmingham, Birmingham, Alabama, United States of America, 2 Department of Biostatistics, University of Alabama at Birmingham, Birmingham, Alabama, United States of America Abstract Uterine leiomyomas (or fibroids) are the most common tumors in women of reproductive age. Early studies of two familial cancer syndromes, the multiple cutaneous and uterine leiomyomatosis (MCUL1), and the hereditary leiomyomatosis and renal cell cancer (HLRCC), implicated FH, a gene on chromosome 1q43 encoding the tricarboxylic acid cycle fumarate hydratase enzyme. The role of this metabolic housekeeping gene in tumorigenesis is still a matter of debate and pseudo- hypoxia has been suggested as a pathological mechanism. Inactivating FH mutations have rarely been observed in the nonsyndromic and common form of fibroids; however, loss of heterozygosity across FH appeared as a significant event in the pathogenesis of a subset of these tumors. To assess the role of FH and the linked genes in nonsyndromic uterine fibroids, we explored a two-megabase interval spanning FH in the NIEHS Uterine fibroid study, a cross-sectional study of fibroids in 1152 premenopausal women. Association mapping with a dense set of single nucleotide polymorphisms revealed several peaks of association (p = 1022–8.1025) with the risk and/or growth of fibroids. In particular, genes encoding factors suspected (cytosolic FH) or known (EXO1 - exonuclease 1) to be involved in DNA mismatch repair emerged as candidate susceptibility genes whereas those acting in the autophagy/apoptosis (MAP1LC3C - microtubule-associated protein) or signal transduction (RGS7 - Regulator of G-protein and PLD5– Phospoholipase D) appeared to affect tumor growth. -

1 AGING Supplementary Table 2
SUPPLEMENTARY TABLES Supplementary Table 1. Details of the eight domain chains of KIAA0101. Serial IDENTITY MAX IN COMP- INTERFACE ID POSITION RESOLUTION EXPERIMENT TYPE number START STOP SCORE IDENTITY LEX WITH CAVITY A 4D2G_D 52 - 69 52 69 100 100 2.65 Å PCNA X-RAY DIFFRACTION √ B 4D2G_E 52 - 69 52 69 100 100 2.65 Å PCNA X-RAY DIFFRACTION √ C 6EHT_D 52 - 71 52 71 100 100 3.2Å PCNA X-RAY DIFFRACTION √ D 6EHT_E 52 - 71 52 71 100 100 3.2Å PCNA X-RAY DIFFRACTION √ E 6GWS_D 41-72 41 72 100 100 3.2Å PCNA X-RAY DIFFRACTION √ F 6GWS_E 41-72 41 72 100 100 2.9Å PCNA X-RAY DIFFRACTION √ G 6GWS_F 41-72 41 72 100 100 2.9Å PCNA X-RAY DIFFRACTION √ H 6IIW_B 2-11 2 11 100 100 1.699Å UHRF1 X-RAY DIFFRACTION √ www.aging-us.com 1 AGING Supplementary Table 2. Significantly enriched gene ontology (GO) annotations (cellular components) of KIAA0101 in lung adenocarcinoma (LinkedOmics). Leading Description FDR Leading Edge Gene EdgeNum RAD51, SPC25, CCNB1, BIRC5, NCAPG, ZWINT, MAD2L1, SKA3, NUF2, BUB1B, CENPA, SKA1, AURKB, NEK2, CENPW, HJURP, NDC80, CDCA5, NCAPH, BUB1, ZWILCH, CENPK, KIF2C, AURKA, CENPN, TOP2A, CENPM, PLK1, ERCC6L, CDT1, CHEK1, SPAG5, CENPH, condensed 66 0 SPC24, NUP37, BLM, CENPE, BUB3, CDK2, FANCD2, CENPO, CENPF, BRCA1, DSN1, chromosome MKI67, NCAPG2, H2AFX, HMGB2, SUV39H1, CBX3, TUBG1, KNTC1, PPP1CC, SMC2, BANF1, NCAPD2, SKA2, NUP107, BRCA2, NUP85, ITGB3BP, SYCE2, TOPBP1, DMC1, SMC4, INCENP. RAD51, OIP5, CDK1, SPC25, CCNB1, BIRC5, NCAPG, ZWINT, MAD2L1, SKA3, NUF2, BUB1B, CENPA, SKA1, AURKB, NEK2, ESCO2, CENPW, HJURP, TTK, NDC80, CDCA5, BUB1, ZWILCH, CENPK, KIF2C, AURKA, DSCC1, CENPN, CDCA8, CENPM, PLK1, MCM6, ERCC6L, CDT1, HELLS, CHEK1, SPAG5, CENPH, PCNA, SPC24, CENPI, NUP37, FEN1, chromosomal 94 0 CENPL, BLM, KIF18A, CENPE, MCM4, BUB3, SUV39H2, MCM2, CDK2, PIF1, DNA2, region CENPO, CENPF, CHEK2, DSN1, H2AFX, MCM7, SUV39H1, MTBP, CBX3, RECQL4, KNTC1, PPP1CC, CENPP, CENPQ, PTGES3, NCAPD2, DYNLL1, SKA2, HAT1, NUP107, MCM5, MCM3, MSH2, BRCA2, NUP85, SSB, ITGB3BP, DMC1, INCENP, THOC3, XPO1, APEX1, XRCC5, KIF22, DCLRE1A, SEH1L, XRCC3, NSMCE2, RAD21. -

1 Uganda Genome Resource Enables Insights
Uganda Genome Resource enables insights into population history and genomic discovery in Africa Gurdasani D.*1, Carstensen T.2* , Fatumo S.* 3,4,5, Chen G.*6, Franklin CS.*2, Prado-Martinez J.* 2, Bouman H.* 2, Abascal F. 2 , Haber M. 2, Tachmazidou I. 2, Mathieson I.7, Ekoru K.8, DeGorter MK. 9, Nsubuga RN.8, Finan C. 2, Wheeler E. 2, Chen L.2, Cooper DN.10, Schiffels S.11, Chen Y. 2, Ritchie GRS.2, Pollard MO. 2, Fortune MD. 2, Mentzer AJ.12, Garrison E. 2, Bergström A.2, Hatzikotoulas K.2, Adebowale A. 6, Doumatey A. 4, Elding H. 2, Wain LV.13,14, Ehret G.15,16, Auer PL.17, Kooperberg CL.18 , Reiner AP.19,20, Franceschini N.21, Maher DP.6, Montgomery SB.7,22, Kadie C.23, Widmer C.24, Xue Y.2, Seeley J. 6,3 , Asiki G. 8 , Kamali A. 8, Young EH. 25,2, Pomilla C. 25, Soranzo N. 2,26,27, Zeggini E. 28, Pirie F.29, Morris AP.30,12, Heckerman D.24, Tyler-Smith C2‡, Motala A.29‡ , Rotimi C6‡, Kaleebu P. ‡3,4,8, Barroso I.‡31,2, Sandhu MS.23 ‡ *these authors contributed equally ‡ these authors contributed equally Lead Contact: Manjinder Sandhu [email protected] 1 William Harvey Research Institute, Queen Mary’s University of London, London, UK 2 Wellcome Sanger Institute, Hinxton, Cambridge, UK 3 London School of Hygiene and Tropical Medicine, London, UK 4 Uganda Medical Informatics Centre (UMIC), MRC/UVRI and LSHTM (Uganda Research Unit), Entebbe-Uganda 5 H3Africa Bioinformatics Network (H3ABioNet) Node, Center for Genomics Research and Innovation (CGRI)/ National Biotechnology Development Agency CGRI/NABDA, Abuja, Nigeria 6 Center for Research -

The Role of Semaphorins and Their Receptors in Vascular Development and Cancer
EXPERIMENTAL CELL RESEARCH ] ( ]]]]) ]]]– ]]] Available online at www.sciencedirect.com journal homepage: www.elsevier.com/locate/yexcr Review Article The role of semaphorins and their receptors in vascular development and cancer Chenghua Gua,n, Enrico Giraudob,nn aDepartment of Neurobiology, Harvard Medical School, 220 Longwood Ave, Boston, MA 02115, USA bInstitute for Cancer Research at Candiolo (IRC@C), and Department of Science and Drug Technology, University of Torino, Str. Prov. 142 Km.3,95 10060 Candiolo, Turin, Italy article information abstract Article Chronology: Semaphorins (Semas) are a large family of traditional axon guidance molecules. Through Received 1 February 2013 interactions with their receptors, Plexins and Neuropilins, Semas play critical roles in a Accepted 6 February 2013 continuously growing list of diverse biological systems. In this review, we focus on their function in regulating vascular development. In addition, over the past few years a number of Keywords: findings have shown the crucial role that Semas and their receptors play in the regulation of Semaphorin cancer progression and tumor angiogenesis. In particular, Semas control tumor progression by Plexin directly influencing the behavior of cancer cells or, indirectly, by modulating angiogenesis and Neuropilin the function of other cell types in the tumor microenvironment (i.e., inflammatory cells and Angiogenesis fibroblasts). Some Semas can activate or inhibit tumor progression and angiogenesis, while Cancer others may have the opposite effect depending on specific post-translational modifications. Here Tumor we will also discuss the diverse biological effects of Semas and their receptor complexes on Development cancer progression as well as their impact on the tumor microenvironment. & Vasculature 2013 Elsevier Inc. -

T-Scan: a Genome-Wide Method for the Systematic Discovery of T Cell Epitopes
HHS Public Access Author manuscript Author ManuscriptAuthor Manuscript Author Cell. Author Manuscript Author manuscript; Manuscript Author available in PMC 2020 January 02. Published in final edited form as: Cell. 2019 August 08; 178(4): 1016–1028.e13. doi:10.1016/j.cell.2019.07.009. T-Scan: A Genome-wide Method for the Systematic Discovery of T Cell Epitopes Tomasz Kula1,2, Mohammad H. Dezfulian1,2, Charlotte I. Wang1,2,3, Nouran S. Abdelfattah1,2, Zachary C. Hartman4, Kai W. Wucherpfennig5, Herbert Kim Lyerly6, Stephen J. Elledge1,2,7,* 1Division of Genetics, Department of Medicine, Howard Hughes Medical Institute, Brigham and Women’s Hospital, Boston, MA 02115, USA 2Department of Genetics, Harvard University Medical School, Boston, MA, USA 3Department of Pathology, Massachusetts General Hospital, Boston, MA, USA 4Departments of Surgery and Pathology, Duke University Medical Center, 571 Research Drive, Suite 433, Box 2606, Durham, NC 27710, USA 5Department of Cancer Immunology and Virology, Dana-Farber Cancer Institute, Department of Immunobiology, Harvard Medical School, Boston, MA 02115, USA 6Departments of Surgery, Immunology, and Pathology, Duke University Medical Center, 571 Research Drive, Suite 433, Box 2606, Durham, NC 27710, USA 7Lead Contact SUMMARY T cell recognition of specific antigens mediates protection from pathogens and controls neoplasias, but can also cause autoimmunity. Our knowledge of T cell antigens and their implications for human health is limited by the technical limitations of T cell profiling technologies. Here, we present T-Scan, a high-throughput platform for identification of antigens productively recognized by T cells. T-Scan uses lentiviral delivery of antigen libraries into cells for endogenous processing and presentation on major histocompatibility complex (MHC) molecules. -

Plexin A4 (PLXNA4) (NM 181775) Human Recombinant Protein – TP761981 | Origene
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for TP761981 Plexin A4 (PLXNA4) (NM_181775) Human Recombinant Protein Product data: Product Type: Recombinant Proteins Description: Purified recombinant protein of Human plexin A4 (PLXNA4), transcript variant 2,full length, with N-terminal GST and C-terminal His tag, expressed in E. coli, 50ug Species: Human Expression Host: E. coli Tag: N-GST and C-His Predicted MW: 86 kDa Concentration: >50 ug/mL as determined by microplate BCA method Purity: > 80% as determined by SDS-PAGE and Coomassie blue staining Buffer: 25mM Tris, pH8.0, 150 mM NaCl, 10% glycerol,1% Sarkosyl. Storage: Store at -80°C. Stability: Stable for 12 months from the date of receipt of the product under proper storage and handling conditions. Avoid repeated freeze-thaw cycles. RefSeq: NP_861440 Locus ID: 91584 UniProt ID: Q9HCM2 RefSeq Size: 2020 Cytogenetics: 7q32.3 RefSeq ORF: 1566 Synonyms: FAYV2820; PLEXA4; PLXNA4A; PLXNA4B; PRO34003 Summary: Coreceptor for SEMA3A. Necessary for signaling by class 3 semaphorins and subsequent remodeling of the cytoskeleton. Plays a role in axon guidance in the developing nervous system. Class 3 semaphorins bind to a complex composed of a neuropilin and a plexin. The plexin modulates the affinity of the complex for specific semaphorins, and its cytoplasmic domain is required for the activation of down-stream signaling events in the cytoplasm (By similarity).[UniProtKB/Swiss-Prot Function] Protein Families: Druggable Genome This product is to be used for laboratory only.