Loss of the Mammalian APC/C Activator FZR1 Shortens G1 and Lengthens S Phase but Has Little Effect on Exit from Mitosis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

PARSANA-DISSERTATION-2020.Pdf
DECIPHERING TRANSCRIPTIONAL PATTERNS OF GENE REGULATION: A COMPUTATIONAL APPROACH by Princy Parsana A dissertation submitted to The Johns Hopkins University in conformity with the requirements for the degree of Doctor of Philosophy Baltimore, Maryland July, 2020 © 2020 Princy Parsana All rights reserved Abstract With rapid advancements in sequencing technology, we now have the ability to sequence the entire human genome, and to quantify expression of tens of thousands of genes from hundreds of individuals. This provides an extraordinary opportunity to learn phenotype relevant genomic patterns that can improve our understanding of molecular and cellular processes underlying a trait. The high dimensional nature of genomic data presents a range of computational and statistical challenges. This dissertation presents a compilation of projects that were driven by the motivation to efficiently capture gene regulatory patterns in the human transcriptome, while addressing statistical and computational challenges that accompany this data. We attempt to address two major difficulties in this domain: a) artifacts and noise in transcriptomic data, andb) limited statistical power. First, we present our work on investigating the effect of artifactual variation in gene expression data and its impact on trans-eQTL discovery. Here we performed an in-depth analysis of diverse pre-recorded covariates and latent confounders to understand their contribution to heterogeneity in gene expression measurements. Next, we discovered 673 trans-eQTLs across 16 human tissues using v6 data from the Genotype Tissue Expression (GTEx) project. Finally, we characterized two trait-associated trans-eQTLs; one in Skeletal Muscle and another in Thyroid. Second, we present a principal component based residualization method to correct gene expression measurements prior to reconstruction of co-expression networks. -

By Submitted in Partial Satisfaction of the Requirements for Degree of in In
Developments of Two Imaging based Technologies for Cell Biology Researches by Xiaowei Yan DISSERTATION Submitted in partial satisfaction of the requirements for degree of DOCTOR OF PHILOSOPHY in Biochemistry and Molecular Biology in the GRADUATE DIVISION of the UNIVERSITY OF CALIFORNIA, SAN FRANCISCO Approved: ______________________________________________________________________________Ronald Vale Chair ______________________________________________________________________________Jonathan Weissman ______________________________________________________________________________Orion Weiner ______________________________________________________________________________ ______________________________________________________________________________ Committee Members Copyright 2021 By Xiaowei Yan ii DEDICATION Everything happens for the best. To my family, who supported me with all their love. iii ACKNOWLEDGEMENTS The greatest joy of my PhD has been joining UCSF, working and learning with such a fantastic group of scientists. I am extremely grateful for all the support and mentorship I received and would like to thank: My mentor, Ron Vale, who is such a great and generous person. Thank you for showing me that science is so much fun and thank you for always giving me the freedom in pursuing my interest. I am grateful for all the guidance from you and thank you for always supporting me whenever I needed. You are a person full of wisdom, and I have been learning so much from you and your attitude to science, science community and even life will continue inspire me. Thank you for being my mentor and thank you for being such a great mentor. Everyone else in Vale lab, past and present, for making our lab a sweet home. I would like to give my special thank to Marvin (Marvin Tanenbaum) and Nico (Nico Stuurman), two other mentors for me in the lab. I would like to thank them for helping me adapt to our lab, for all the valuable advice and for all the happiness during the time that we work together. -
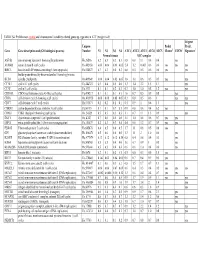
TABLE S4: Proliferation-Related and Chromosomal Instability-Related
TABLE S4: Proliferation-related and chromosomal instability-related genes up-regulated in ATC (weight <4.0) 44-gene Unigene Prolif. Prolif. Gene Gene description and (GO biological process) Number N1 N2 N3 N4 ATC1 ATC2 ATC3 ATC4 ATC5 Cluster1 CIN702 Signature3 Normal tissues ATC samples ASF1B anti-silencing function 1 homolog B (unknown) Hs.26516 0.2 0.3 0.3 0.3 0.8 0.8 1.1 0.8 0.4 yes AURKB aurora kinase B (cell cycle) Hs.442658 0.06 0.06 0.08 0.05 2.4 1.2 0.003 0.6 0.6 yes yes yes BIRC5 baculoviral IAP repeat-containing 5 (anti apoptosis) Hs.514527 0.7 0.3 0.4 0.3 0.8 0.5 0.5 0.6 0.6 yes yes budding uninhibited by benzimidazoles 1 homolog (mitotic BUB1 spindle checkpoint) Hs.469649 0.06 0.04 0.02 0.05 1.8 1.0 0.6 0.5 0.5 yes yes CCNE1 cyclin E1 (cell cycle) Hs.244723 0.5 0.4 0.5 0.6 1.3 1.4 2.2 1.5 1.1 yes CCNF cyclin F (cell cycle) Hs.1973 0.1 0.1 0.2 0.3 0.7 1.0 1.0 0.8 1.2 yes yes CDC45L CDC45 cell division cycle 45-like (cell cycle) Hs.474217 0.1 0.1 0.1 0.3 1.6 0.7 0.5 0.9 0.8 yes CDC6 cell division cycle 6 homolog (cell cycle) Hs. 405958 0.08 0.08 0.08 0.07 0.3 0.8 0.5 0.6 1 yes yes CDC7 cell division cycle 7 (cell cycle) Hs.533573 0.2 0.2 0.2 0.1 0.5 0.9 1 0.6 1.3 yes CDKN3 cyclin-dependent kinase inhibitor 3 (cell cycle) Hs.84113 0.1 0.1 0.7 0.1 0.9 0.8 0.6 0.4 6.2 yes CHEK1 CHK1 checkpoint homolog (cell cycle) Hs. -

Predicting FOXM1-Mediated Gene Regulation Through the Analysis of Genome-Wide FOXM1 Binding Sites in MCF-7, K562, SK-N-SH, GM12878 and ECC-1 Cell Lines
International Journal of Molecular Sciences Article Predicting FOXM1-Mediated Gene Regulation through the Analysis of Genome-Wide FOXM1 Binding Sites in MCF-7, K562, SK-N-SH, GM12878 and ECC-1 Cell Lines 1, , 2, 1 3,4 Keunsoo Kang * y, Yoonjung Choi y, Hoo Hyun Kim , Kyung Hyun Yoo and Sungryul Yu 5,* 1 Department of Microbiology, College of Science & Technology, Dankook University, Cheonan 31116, Korea; [email protected] 2 Deargen Inc., Daejeon 34051, Korea; [email protected] 3 Laboratory of Biomedical Genomics, Department of Biological Sciences, Sookmyung Women’s University, Seoul 04310, Korea; [email protected] 4 Research Institute of Women’s Health, Sookmyung Women’s University, Seoul 04310, Korea 5 Department of Clinical Laboratory Science, Semyung University, Jecheon 27136, Korea * Correspondence: [email protected] (K.K.); [email protected] (S.Y.); Tel.: +82-41-550-3456 (K.K.); +82-43-649-1418 (S.Y.) These authors contributed equally to the work. y Received: 22 June 2020; Accepted: 24 August 2020; Published: 26 August 2020 Abstract: Forkhead box protein M1 (FOXM1) is a key transcription factor (TF) that regulates a common set of genes related to the cell cycle in various cell types. However, the mechanism by which FOXM1 controls the common gene set in different cellular contexts is unclear. In this study, a comprehensive meta-analysis of genome-wide FOXM1 binding sites in ECC-1, GM12878, K562, MCF-7, and SK-N-SH cell lines was conducted to predict FOXM1-driven gene regulation. Consistent with previous studies, different TF binding motifs were identified at FOXM1 binding sites, while the NFY binding motif was found at 81% of common FOXM1 binding sites in promoters of cell cycle-related genes. -

1 AGING Supplementary Table 2
SUPPLEMENTARY TABLES Supplementary Table 1. Details of the eight domain chains of KIAA0101. Serial IDENTITY MAX IN COMP- INTERFACE ID POSITION RESOLUTION EXPERIMENT TYPE number START STOP SCORE IDENTITY LEX WITH CAVITY A 4D2G_D 52 - 69 52 69 100 100 2.65 Å PCNA X-RAY DIFFRACTION √ B 4D2G_E 52 - 69 52 69 100 100 2.65 Å PCNA X-RAY DIFFRACTION √ C 6EHT_D 52 - 71 52 71 100 100 3.2Å PCNA X-RAY DIFFRACTION √ D 6EHT_E 52 - 71 52 71 100 100 3.2Å PCNA X-RAY DIFFRACTION √ E 6GWS_D 41-72 41 72 100 100 3.2Å PCNA X-RAY DIFFRACTION √ F 6GWS_E 41-72 41 72 100 100 2.9Å PCNA X-RAY DIFFRACTION √ G 6GWS_F 41-72 41 72 100 100 2.9Å PCNA X-RAY DIFFRACTION √ H 6IIW_B 2-11 2 11 100 100 1.699Å UHRF1 X-RAY DIFFRACTION √ www.aging-us.com 1 AGING Supplementary Table 2. Significantly enriched gene ontology (GO) annotations (cellular components) of KIAA0101 in lung adenocarcinoma (LinkedOmics). Leading Description FDR Leading Edge Gene EdgeNum RAD51, SPC25, CCNB1, BIRC5, NCAPG, ZWINT, MAD2L1, SKA3, NUF2, BUB1B, CENPA, SKA1, AURKB, NEK2, CENPW, HJURP, NDC80, CDCA5, NCAPH, BUB1, ZWILCH, CENPK, KIF2C, AURKA, CENPN, TOP2A, CENPM, PLK1, ERCC6L, CDT1, CHEK1, SPAG5, CENPH, condensed 66 0 SPC24, NUP37, BLM, CENPE, BUB3, CDK2, FANCD2, CENPO, CENPF, BRCA1, DSN1, chromosome MKI67, NCAPG2, H2AFX, HMGB2, SUV39H1, CBX3, TUBG1, KNTC1, PPP1CC, SMC2, BANF1, NCAPD2, SKA2, NUP107, BRCA2, NUP85, ITGB3BP, SYCE2, TOPBP1, DMC1, SMC4, INCENP. RAD51, OIP5, CDK1, SPC25, CCNB1, BIRC5, NCAPG, ZWINT, MAD2L1, SKA3, NUF2, BUB1B, CENPA, SKA1, AURKB, NEK2, ESCO2, CENPW, HJURP, TTK, NDC80, CDCA5, BUB1, ZWILCH, CENPK, KIF2C, AURKA, DSCC1, CENPN, CDCA8, CENPM, PLK1, MCM6, ERCC6L, CDT1, HELLS, CHEK1, SPAG5, CENPH, PCNA, SPC24, CENPI, NUP37, FEN1, chromosomal 94 0 CENPL, BLM, KIF18A, CENPE, MCM4, BUB3, SUV39H2, MCM2, CDK2, PIF1, DNA2, region CENPO, CENPF, CHEK2, DSN1, H2AFX, MCM7, SUV39H1, MTBP, CBX3, RECQL4, KNTC1, PPP1CC, CENPP, CENPQ, PTGES3, NCAPD2, DYNLL1, SKA2, HAT1, NUP107, MCM5, MCM3, MSH2, BRCA2, NUP85, SSB, ITGB3BP, DMC1, INCENP, THOC3, XPO1, APEX1, XRCC5, KIF22, DCLRE1A, SEH1L, XRCC3, NSMCE2, RAD21. -

Role of Fzr1 in Embryogenesis
ROLE OF FZR1 IN EMBRYOGENESIS SEAH KAY YI MICHELLE BSC. (HONS I) PH.D THESIS Statement of Originality This thesis contains no material which has been accepted for the award for any other Degree or Diploma in any University or other tertiary institution and, to the best of my knowledge and belief, contains no material previously published or written by another person, except where due reference has been made in the text. I give consent to this copy of my thesis, when deposited in the University library, being made available for loan and photocopying subject to the provisions of the Copyright Act 1968. Seah Kay Yi Michelle 17th December 2012 Page | ii Acknowledgements I would like to sincerely express my appreciation and gratitude to my supervisors, Keith and Janet for giving me this opportunity and to share their wisdom and guidance throughout my PhD. I would also like to extend my thanks to all the lab members including Evan, Jess, Julie, Kyra, Nicole, Phoebe, Simon, Sophia, Suzanne and Yan. Thank you for your company and help throughout my PhD, it has made this an enjoyable experience in the lab. To my family especially Popo, Mummy and Daddy, thank you for the endless love, support and understanding that you have showered upon me. For that, I will be eternally grateful. Thank you for moulding me into the person that I am today, for without all of you, I will not be where I am today. I love you and will always be your little girl. To Clara, my BFF, thank you for your encouragements and to always be there for me. -

Emi1 (FBXO5) (Center) Rabbit Polyclonal Antibody Product Data
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for AP51634PU-N Emi1 (FBXO5) (Center) Rabbit Polyclonal Antibody Product data: Product Type: Primary Antibodies Applications: WB Recommended Dilution: ELISA: 1/1000. Western Blot: 1/100-1/500. Reactivity: Human Host: Rabbit Isotype: Ig Clonality: Polyclonal Immunogen: KLH conjugated synthetic peptide between 191-221 amino acids from the Central region of Human FBXO5. Specificity: This antibody recognizes Human FBXO5 (Center). Formulation: PBS containing 0.09% (W/V) Sodium Azide as preservative State: Aff - Purified State: Liquid purified Ig fraction Concentration: lot specific Purification: Affinity Chromatography on Protein A Conjugation: Unconjugated Storage: Store undiluted at 2-8°C for one month or (in aliquots) at -20°C for longer. Avoid repeated freezing and thawing. Stability: Shelf life: one year from despatch. Gene Name: Homo sapiens F-box protein 5 (FBXO5), transcript variant 2 Database Link: Entrez Gene 26271 Human Q9UKT4 This product is to be used for laboratory only. Not for diagnostic or therapeutic use. View online » ©2021 OriGene Technologies, Inc., 9620 Medical Center Drive, Ste 200, Rockville, MD 20850, US 1 / 2 Emi1 (FBXO5) (Center) Rabbit Polyclonal Antibody – AP51634PU-N Background: This gene encodes a member of the F-box protein family which is characterized by an approximately 40 amino acid motif, the F-box. The F-box proteins constitute one of the four subunits of the ubiquitin protein ligase complex called SCFs (SKP1-cullin-F-box), which function in phosphorylation-dependent ubiquitination. -

Human Lectins, Their Carbohydrate Affinities and Where to Find Them
biomolecules Review Human Lectins, Their Carbohydrate Affinities and Where to Review HumanFind Them Lectins, Their Carbohydrate Affinities and Where to FindCláudia ThemD. Raposo 1,*, André B. Canelas 2 and M. Teresa Barros 1 1, 2 1 Cláudia D. Raposo * , Andr1 é LAQVB. Canelas‐Requimte,and Department M. Teresa of Chemistry, Barros NOVA School of Science and Technology, Universidade NOVA de Lisboa, 2829‐516 Caparica, Portugal; [email protected] 12 GlanbiaLAQV-Requimte,‐AgriChemWhey, Department Lisheen of Chemistry, Mine, Killoran, NOVA Moyne, School E41 of ScienceR622 Co. and Tipperary, Technology, Ireland; canelas‐ [email protected] NOVA de Lisboa, 2829-516 Caparica, Portugal; [email protected] 2* Correspondence:Glanbia-AgriChemWhey, [email protected]; Lisheen Mine, Tel.: Killoran, +351‐212948550 Moyne, E41 R622 Tipperary, Ireland; [email protected] * Correspondence: [email protected]; Tel.: +351-212948550 Abstract: Lectins are a class of proteins responsible for several biological roles such as cell‐cell in‐ Abstract:teractions,Lectins signaling are pathways, a class of and proteins several responsible innate immune for several responses biological against roles pathogens. such as Since cell-cell lec‐ interactions,tins are able signalingto bind to pathways, carbohydrates, and several they can innate be a immuneviable target responses for targeted against drug pathogens. delivery Since sys‐ lectinstems. In are fact, able several to bind lectins to carbohydrates, were approved they by canFood be and a viable Drug targetAdministration for targeted for drugthat purpose. delivery systems.Information In fact, about several specific lectins carbohydrate were approved recognition by Food by andlectin Drug receptors Administration was gathered for that herein, purpose. plus Informationthe specific organs about specific where those carbohydrate lectins can recognition be found by within lectin the receptors human was body. -

Differential Gene Expression in the Skin of Xiphophorus
DIFFERENTIAL GENE EXPRESSION IN THE SKIN OF XIPHOPHORUS MACULATUS JP 163 B IN RESPONSE TO FULL SPECTRUM (10,000 K) FLUORESCENT LIGHT by Kaela Caballero, B.S. A thesis submitted to the Graduate Council of Texas State University in partial fulfillment of the requirements for the degree of Master of Science with a Major in Biochemistry August 2015 Committee Members: Ronald Walter, Chair Rachell Booth Steven Whitten COPYRIGHT by Kaela L. Caballero 2015 FAIR USE AND AUTHOR’S PERMISSION STATEMENT Fair Use This work is protected by the Copyright Laws of the United States (Public Law 94-553, section 107). Consistent with fair use as defined in the Copyright Laws, brief quotations from this material are allowed with proper acknowledgment. Use of this material for financial gain without the author’s express written permission is not allowed. Duplication Permission As the copyright holder of this work I, Kaela L. Caballero, authorize duplication of this work, in whole or in part, for educational or scholarly purposes only. DEDICATION To my parents and family, who have always supported and encouraged me, my education and my coffee addiction. And to Max, my study buddy; where you have gone, there is no need for coffee fueled late nights. Rest in peace. ACKNOWLEDGEMENTS This work could not have been accomplished without the support and mentorship that I have received throughout my life from family, teachers, mentors and friends. It is impossible to name everyone here, but I would like to use a few lines to thank those that have helped me achieve my goals. Thank you to Dr. -

Expression of a Mirna Targeting Mutated SOD1 in Astrocytes Induces Motoneuron Plasticity and Improves Neuromuscular Function in ALS Mice
bioRxiv preprint doi: https://doi.org/10.1101/2021.01.08.425706; this version posted January 9, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Expression of a miRNA targeting mutated SOD1 in astrocytes induces motoneuron plasticity and improves neuromuscular function in ALS mice Rochat C.1, Bernard-Marissal N.1,6, Pradervand S.3, Perrin F.E.4, Raoul C.5, Aebischer P.1, Schneider B.L.1,2* 1 Brain Mind Institute, Ecole Polytechnique Fédérale de Lausanne (EPFL), 1015 Lausanne, Switzerland 2 Bertarelli Platform for Gene Therapy, Ecole Polytechnique Fédérale de Lausanne (EPFL), Geneva, Switzerland 3 Genomic Technologies Facility, University of Lausanne, Lausanne, Switzerland. 4 INSERM U1198, University of Montpellier, EPHE, Place Eugène Bataillon CC105, F-34095, Montpellier, France 5 The Neuroscience Institute of Montpellier, Inserm UMR1051, Univ Montpellier, Saint Eloi Hospital, Montpellier, France 6 Aix Marseille Univ, INSERM, MMG, Marseille, France *Correspondence should be addressed to B.L.S. ([email protected]) Short title: miRNA gene therapy targeting astrocytes in ALS Study was performed in Lausanne, Switzerland Current address of corresponding author: Bernard Schneider EPFL SV PTECH PTBTG Ch. des Mines 9 CH-1202 Genève Switzerland Email: [email protected] Tel: +41 21 693 95 05 bioRxiv preprint doi: https://doi.org/10.1101/2021.01.08.425706; this version posted January 9, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. -

Comparison of Gene Expression Response to Neutron and X-Ray Irradiation Using Mouse Blood Constantinos G
Broustas et al. BMC Genomics (2017) 18:2 DOI 10.1186/s12864-016-3436-1 RESEARCHARTICLE Open Access Comparison of gene expression response to neutron and x-ray irradiation using mouse blood Constantinos G. Broustas1, Yanping Xu2, Andrew D. Harken2, Guy Garty2 and Sally A. Amundson1* Abstract Background: In the event of an improvised nuclear device detonation, the prompt radiation exposure would consist of photons plus a neutron component that would contribute to the total dose. As neutrons cause more complex and difficult to repair damage to cells that would result in a more severe health burden to affected individuals, it is paramount to be able to estimate the contribution of neutrons to an estimated dose, to provide information for those making treatment decisions. Results: Mice exposed to either 0.25 or 1 Gy of neutron or 1 or 4 Gy x-ray radiation were sacrificed at 1 or 7 days after exposure. Whole genome microarray analysis identified 7285 and 5045 differentially expressed genes in the blood of mice exposed to neutron or x-ray radiation, respectively. Neutron exposure resulted in mostly downregulated genes, whereas x-rays showed both down- and up-regulated genes. A total of 34 differentially expressed genes were regulated in response to all ≥1 Gy exposures at both times. Of these, 25 genes were consistently downregulated at days 1 and 7, whereas 9 genes, including the transcription factor E2f2, showed bi-directional regulation; being downregulated at day 1, while upregulated at day 7. Gene ontology analysis revealed that genes involved in nucleic acid metabolism processes were persistently downregulated in neutron irradiated mice, whereas genes involved in lipid metabolism were upregulated in x-ray irradiated animals. -

Phosphorylation of the Anaphase Promoting Complex
www.nature.com/scientificreports OPEN Phosphorylation of the Anaphase Promoting Complex activator FZR1/CDH1 is required for Meiosis II entry in mouse male germ cell Nobuhiro Tanno1,2, Shinji Kuninaka2, Sayoko Fujimura3, Kazumasa Takemoto1, Kaho Okamura1, Naoki Takeda4, Kimi Araki4,5, Masatake Araki4, Hideyuki Saya2 & Kei-ichiro Ishiguro1 ✉ FZR1/CDH1 is an activator of Anaphase promoting complex/Cyclosome (APC/C), best known for its role as E3 ubiquitin ligase that drives the cell cycle. APC/C activity is regulated by CDK-mediated phosphorylation of FZR1 during mitotic cell cycle. Although the critical role of FZR1 phosphorylation has been shown mainly in yeast and in vitro cell culture studies, its biological signifcance in mammalian tissues in vivo remained elusive. Here, we examined the in vivo role of FZR1 phosphorylation using a mouse model, in which non-phosphorylatable substitutions were introduced in the putative CDK- phosphorylation sites of FZR1. Although ablation of FZR1 phosphorylation did not show substantial consequences in mouse somatic tissues, it led to severe testicular defects resulting in male infertility. In the absence of FZR1 phosphorylation, male juvenile germ cells entered meiosis normally but failed to enter meiosis II or form diferentiated spermatids. In aged testis, male mutant germ cells were overall abolished, showing Sertoli cell-only phenotype. In contrast, female mutants showed apparently normal progression of meiosis. The present study demonstrated that phosphorylation of FZR1 is required for temporal regulation of APC/C activity at meiosis II entry, and for maintenance of spermatogonia, which raised an insight into the sexual dimorphism of FZR1-regulation in germ cells. Anaphase promoting complex/Cyclosome (APC/C) controls timely transitions of mitotic cell cycle phases by pro- moting ubiquitylation and degradation of many key cell cycle regulators1.