Alterations in Keratin Levels and Modifications in Colorectal Adenomagenesis Ria Rosser
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Keratin 19 Regulates Cell Shape and Cell-Cell Adhesion of MCF7 Cells While Maintaining E-Cadherin Localization at the Cell Surface
Keratin 19 regulates cell shape and cell-cell adhesion of MCF7 cells while maintaining E-cadherin localization at the cell surface Welcome to my poster. This is Sarah Alsharif, a PhD student from the biology department. I am glad to present the work our lab has been doing in the breast cancer field. In fact, after lung cancer, breast cancer is the second cause of death in women worldwide (1). It is estimated that every 18 seconds, approximately one new case of breast cancer is documented (2). No one dies due to cancer itself. The death is because of metastasis which takes place when cancer cells leave a breast in which they are formed and reach other sites such as the brain or lung. Our lab is interested in investigating the mechanism behind metastasis of breast cancer. Metastasis is associated with what is called epithelial to mesenchymal transition (EMT), the process characterized by loss of cell to cell adhesion and expression of epithelial markers such as keratin intermediate filament proteins, as you can see in the first three images of cells. Those filaments are keratins and they are critical for the shape and for maintaining mechanical integrity of epithelial cells via cell to cell complexes called desmosomes. Among different keratins, keratin 19 (K19) is highly expressed in many types of cancer including breast cancer, and is correlated with a worse prognosis (3). Consistently, K19 expression has been reported to be significantly higher in metastatic breast cancer tumor cells compared to primary tumors (4). The role of K19 on mechanical properties of cancer cells for cell migration and possible impact on metastasis in breast cancer patients is still unknown. -

Genetic Background Effects of Keratin 8 and 18 in a DDC-Induced Hepatotoxicity and Mallory-Denk Body Formation Mouse Model
Laboratory Investigation (2012) 92, 857–867 & 2012 USCAP, Inc All rights reserved 0023-6837/12 $32.00 Genetic background effects of keratin 8 and 18 in a DDC-induced hepatotoxicity and Mallory-Denk body formation mouse model Johannes Haybaeck1, Cornelia Stumptner1, Andrea Thueringer1, Thomas Kolbe2, Thomas M Magin3, Michael Hesse4, Peter Fickert5, Oleksiy Tsybrovskyy1, Heimo Mu¨ller1, Michael Trauner5,6, Kurt Zatloukal1 and Helmut Denk1 Keratin 8 (K8) and keratin 18 (K18) form the major hepatocyte cytoskeleton. We investigated the impact of genetic loss of either K8 or K18 on liver homeostasis under toxic stress with the hypothesis that K8 and K18 exert different functions. krt8À/À and krt18À/À mice crossed into the same 129-ola genetic background were treated by acute and chronic ad- ministration of 3,5-diethoxy-carbonyl-1,4-dihydrocollidine (DDC). In acutely DDC-intoxicated mice, macrovesicular steatosis was more pronounced in krt8À/À and krt18À/À compared with wild-type (wt) animals. Mallory-Denk bodies (MDBs) appeared in krt18À/À mice already at an early stage of intoxication in contrast to krt8À/À mice that did not display MDB formation when fed with DDC. Keratin-deficient mice displayed significantly lower numbers of apoptotic hepatocytes than wt animals. krt8À/À, krt18À/À and control mice displayed comparable cell proliferation rates. Chronically DDC-intoxicated krt18À/À and wt mice showed a similarly increased degree of steatohepatitis with hepatocyte ballooning and MDB formation. In krt8À/À mice, steatosis was less, ballooning, and MDBs were absent. krt18À/À mice developed MDBs whereas krt8À/À mice on the same genetic background did not, highlighting the significance of different structural properties of keratins. -

Appropriate Roles of Cardiac Troponins in Evaluating Patients with Chest Pain
J Am Board Fam Pract: first published as 10.3122/jabfm.12.3.214 on 1 May 1999. Downloaded from MEDICAL PRACTICE Appropriate Roles of Cardiac Troponins in Evaluating Patients With Chest Pain Matthew S. Rice, MD, CPT, Me, USA, and David C. MacDonald, DO, Me, USA Background: Diagnosis of acute myocardial infarction relies upon the clinical history, interpretation of the electrocardiogram, and measurement of serum levels of cardiac enzymes. Newer biochemical markers of myocardial injury, such as cardiac troponin I and cardiac troponin T, are now being used instead of or along with the standard markers, the MB isoenzyme of creatine kinase (CK-MB) and lactate dehydrogenase. Methods: We performed a MEDLINE literature search (1987 to 1997) using the key words "troponin I," "troponin T," and "acute myocardial infarction." We reviewed selected articles related to the diagnostic and prognostic usefulness of these cardiac markers in evaluating patients with suspected myocardial infarction. Results: We found that (1) troponin I is a better cardiac marker than CK-MB for myocardial infarction because it is equally sensitive yet more specific for myocardial injury; (2) troponin T is a relatively poorer cardiac marker than CK-MB because it is less sensitive and less specific for myocardial injury; and (3) both troponin I and troponin T may be used as independent prognosticators of future cardiac events. Conclusions: Troponin I is a sensitive and specific marker for myocardial injury and can be used to predict the likelihood of future cardiac events. It is not much more expensive to measure than CK-MB. Over all, troponin I is a better cardiac marker than CK-MB and should become the preferred cardiac enzyme when evaluating patients with suspected myocardial infarction. -

Quantigene Flowrna Probe Sets Currently Available
QuantiGene FlowRNA Probe Sets Currently Available Accession No. Species Symbol Gene Name Catalog No. NM_003452 Human ZNF189 zinc finger protein 189 VA1-10009 NM_000057 Human BLM Bloom syndrome VA1-10010 NM_005269 Human GLI glioma-associated oncogene homolog (zinc finger protein) VA1-10011 NM_002614 Human PDZK1 PDZ domain containing 1 VA1-10015 NM_003225 Human TFF1 Trefoil factor 1 (breast cancer, estrogen-inducible sequence expressed in) VA1-10016 NM_002276 Human KRT19 keratin 19 VA1-10022 NM_002659 Human PLAUR plasminogen activator, urokinase receptor VA1-10025 NM_017669 Human ERCC6L excision repair cross-complementing rodent repair deficiency, complementation group 6-like VA1-10029 NM_017699 Human SIDT1 SID1 transmembrane family, member 1 VA1-10032 NM_000077 Human CDKN2A cyclin-dependent kinase inhibitor 2A (melanoma, p16, inhibits CDK4) VA1-10040 NM_003150 Human STAT3 signal transducer and activator of transcripton 3 (acute-phase response factor) VA1-10046 NM_004707 Human ATG12 ATG12 autophagy related 12 homolog (S. cerevisiae) VA1-10047 NM_000737 Human CGB chorionic gonadotropin, beta polypeptide VA1-10048 NM_001017420 Human ESCO2 establishment of cohesion 1 homolog 2 (S. cerevisiae) VA1-10050 NM_197978 Human HEMGN hemogen VA1-10051 NM_001738 Human CA1 Carbonic anhydrase I VA1-10052 NM_000184 Human HBG2 Hemoglobin, gamma G VA1-10053 NM_005330 Human HBE1 Hemoglobin, epsilon 1 VA1-10054 NR_003367 Human PVT1 Pvt1 oncogene homolog (mouse) VA1-10061 NM_000454 Human SOD1 Superoxide dismutase 1, soluble (amyotrophic lateral sclerosis 1 (adult)) -

Identification of Trichoplein, a Novel Keratin Filament- Binding Protein
Research Article 1081 Identification of trichoplein, a novel keratin filament- binding protein Miwako Nishizawa1,*, Ichiro Izawa1,*, Akihito Inoko1,*, Yuko Hayashi1, Koh-ichi Nagata1, Tomoya Yokoyama1,2, Jiro Usukura3 and Masaki Inagaki1,‡ 1Division of Biochemistry, Aichi Cancer Center Research Institute, 1-1 Kanokoden, Chikusa-ku, Nagoya 464-8681, Japan 2Department of Dermatology, Mie University Faculty of Medicine, 2-174 Edobashi, Tsu 514-8507, Japan 3Department of Anatomy and Cell Biology, Nagoya University School of Medicine, 65 Tsurumai, Showa-ku, Nagoya 466-8550, Japan *These authors contributed equally to this work ‡Author for correspondence (e-mail: [email protected]) Accepted 29 November 2004 Journal of Cell Science 118, 1081-1090 Published by The Company of Biologists 2005 doi:10.1242/jcs.01667 Summary Keratins 8 and 18 (K8/18) are major components of the antibody in a complex with K8/18 and immunostaining intermediate filaments (IFs) of simple epithelia. We report revealed that trichoplein colocalized with K8/18 filaments here the identification of a novel protein termed in HeLa cells. In polarized Caco-2 cells, trichoplein trichoplein. This protein shows a low degree of sequence colocalized not only with K8/18 filaments in the apical similarity to trichohyalin, plectin and myosin heavy chain, region but also with desmoplakin, a constituent of and is a K8/18-binding protein. Among interactions desmosomes. In the absorptive cells of the small intestine, between trichoplein and various IF proteins that we trichoplein colocalized with K8/18 filaments at the apical tested using two-hybrid methods, trichoplein interacted cortical region, and was also concentrated at desmosomes. -
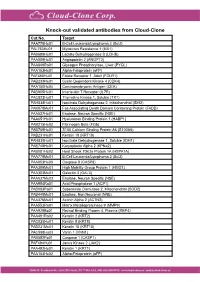
Knock-Out Validated Antibodies from Cloud-Clone Cat.No
Knock-out validated antibodies from Cloud-Clone Cat.No. Target PAA778Hu01 B-Cell Leukemia/Lymphoma 2 (Bcl2) PAL763Hu01 Myxovirus Resistance 1 (MX1) PAB698Hu01 Lactate Dehydrogenase B (LDHB) PAA009Hu01 Angiopoietin 2 (ANGPT2) PAA849Ra01 Glycogen Phosphorylase, Liver (PYGL) PAA153Hu01 Alpha-Fetoprotein (aFP) PAF460Hu01 Folate Receptor 1, Adult (FOLR1) PAB233Hu01 Cyclin Dependent Kinase 4 (CDK4) PAA150Hu04 Carcinoembryonic Antigen (CEA) PAB905Hu01 Interleukin 7 Receptor (IL7R) PAC823Hu01 Thymidine Kinase 1, Soluble (TK1) PAH838Hu01 Isocitrate Dehydrogenase 2, mitochondrial (IDH2) PAK078Mu01 Fas Associating Death Domain Containing Protein (FADD) PAA537Hu01 Enolase, Neuron Specific (NSE) PAA651Hu01 Hyaluronan Binding Protein 1 (HABP1) PAB215Hu02 Fibrinogen Beta (FGb) PAB769Hu01 S100 Calcium Binding Protein A6 (S100A6) PAB231Hu01 Keratin 18 (KRT18) PAH839Hu01 Isocitrate Dehydrogenase 1, Soluble (IDH1) PAE748Hu01 Karyopherin Alpha 2 (KPNa2) PAB081Hu02 Heat Shock 70kDa Protein 1A (HSPA1A) PAA778Mu01 B-Cell Leukemia/Lymphoma 2 (Bcl2) PAA853Hu03 Caspase 8 (CASP8) PAA399Mu01 High Mobility Group Protein 1 (HMG1) PAA303Mu01 Galectin 3 (GAL3) PAA537Mu02 Enolase, Neuron Specific (NSE) PAA994Ra01 Acid Phosphatase 1 (ACP1) PAB083Ra01 Superoxide Dismutase 2, Mitochondrial (SOD2) PAB449Mu01 Enolase, Non Neuronal (NNE) PAA376Mu01 Actinin Alpha 2 (ACTN2) PAA553Ra01 Matrix Metalloproteinase 9 (MMP9) PAA929Bo01 Retinol Binding Protein 4, Plasma (RBP4) PAA491Ra02 Keratin 2 (KRT2) PAC025Hu01 Keratin 8 (KRT8) PAB231Mu01 Keratin 18 (KRT18) PAC598Hu03 Vanin 1 (VNN1) -

Gene Therapy Rescues Cardiac Dysfunction in Duchenne Muscular
JACC: BASIC TO TRANSLATIONAL SCIENCE VOL.4,NO.7,2019 ª 2019 THE AUTHORS. PUBLISHED BY ELSEVIER ON BEHALF OF THE AMERICAN COLLEGE OF CARDIOLOGY FOUNDATION. THIS IS AN OPEN ACCESS ARTICLE UNDER THE CC BY-NC-ND LICENSE (http://creativecommons.org/licenses/by-nc-nd/4.0/). PRECLINICAL RESEARCH Gene Therapy Rescues Cardiac DysfunctioninDuchenneMuscular Dystrophy Mice by Elevating Cardiomyocyte Deoxy-Adenosine Triphosphate a b c d,e Stephen C. Kolwicz, JR,PHD, John K. Hall, PHD, Farid Moussavi-Harami, MD, Xiolan Chen, PHD, d,e b,d,e e,f,g, b,e,g, Stephen D. Hauschka, PHD, Jeffrey S. Chamberlain, PHD, Michael Regnier, PHD, * Guy L. Odom, PHD * VISUAL ABSTRACT Kolwicz, S.C. Jr. et al. J Am Coll Cardiol Basic Trans Science. 2019;4(7):778–91. HIGHLIGHTS rAAV vectors increase cardiac-specific expression of RNR and elevate cardiomyocyte 2-dATP levels. Elevated myocardial RNR and subsequent increase in 2-dATP rescues the performance of failing myocardium, an effect that persists long term. ISSN 2452-302X https://doi.org/10.1016/j.jacbts.2019.06.006 JACC: BASIC TO TRANSLATIONAL SCIENCE VOL. 4, NO. 7, 2019 Kolwicz, Jr., et al. 779 NOVEMBER 2019:778– 91 Nucleotide-Based Cardiac Gene Therapy Restores Function in dmd Mice We show the ability to increase both cardiac baseline function and high workload contractile performance in ABBREVIATIONS aged (22- to 24-month old) mdx4cv mice, by high-level muscle-specific expression of either microdystrophin AND ACRONYMS or RNR. mDys = microdystrophin Five months post-treatment, mice systemically injected with rAAV6 vector carrying a striated muscle-specific CK8 regulatory cassette driving expression of microdystrophin in both skeletal and cardiac muscle, exhibited the = miniaturized murine creatine kinase regulatory greatest effect on systolic function. -

Hypertrophic Cardiomyopathy- Associated Mutations in Genes That Encode Calcium-Handling Proteins
Current Molecular Medicine 2012, 12, 507-518 507 Beyond the Cardiac Myofilament: Hypertrophic Cardiomyopathy- Associated Mutations in Genes that Encode Calcium-Handling Proteins A.P. Landstrom and M.J. Ackerman* Departments of Medicine, Pediatrics, and Molecular Pharmacology & Experimental Therapeutics, Divisions of Cardiovascular Diseases and Pediatric Cardiology, and the Windland Smith Rice Sudden Death Genomics Laboratory, Mayo Clinic, Rochester, Minnesota, USA Abstract: Traditionally regarded as a genetic disease of the cardiac sarcomere, hypertrophic cardiomyopathy (HCM) is the most common inherited cardiovascular disease and a significant cause of sudden cardiac death. While the most common etiologies of this phenotypically diverse disease lie in a handful of genes encoding critical contractile myofilament proteins, approximately 50% of patients diagnosed with HCM worldwide do not host sarcomeric gene mutations. Recently, mutations in genes encoding calcium-sensitive and calcium- handling proteins have been implicated in the pathogenesis of HCM. Among these are mutations in TNNC1- encoded cardiac troponin C, PLN-encoded phospholamban, and JPH2-encoded junctophilin 2 which have each been associated with HCM in multiple studies. In addition, mutations in RYR2-encoded ryanodine receptor 2, CASQ2-encoded calsequestrin 2, CALR3-encoded calreticulin 3, and SRI-encoded sorcin have been associated with HCM, although more studies are required to validate initial findings. While a relatively uncommon cause of HCM, mutations in genes that encode calcium-handling proteins represent an emerging genetic subset of HCM. Furthermore, these naturally occurring disease-associated mutations have provided useful molecular tools for uncovering novel mechanisms of disease pathogenesis, increasing our understanding of basic cardiac physiology, and dissecting important structure-function relationships within these proteins. -

Keratins Couple with the Nuclear Lamina and Regulate Proliferation in Colonic Epithelial Cells Carl-Gustaf A
bioRxiv preprint doi: https://doi.org/10.1101/2020.06.22.164467; this version posted June 22, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Keratins couple with the nuclear lamina and regulate proliferation in colonic epithelial cells Carl-Gustaf A. Stenvall1*, Joel H. Nyström1*, Ciarán Butler-Hallissey1,5, Stephen A. Adam2, Roland Foisner3, Karen M. Ridge2, Robert D. Goldman2, Diana M. Toivola1,4 1 Cell Biology, Biosciences, Faculty of Science and Engineering, Åbo Akademi University, Turku, Finland 2 Department of Cell and Developmental Biology, Feinberg School of Medicine, Northwestern University, Chicago, Illinois, USA 3 Max Perutz Labs, Medical University of Vienna, Vienna Biocenter Campus (VBC), Vienna, Austria 4 Turku Center for Disease Modeling, Turku, Finland 5 Turku Bioscience Centre, University of Turku and Åbo Akademi University, Turku, Finland * indicates equal contribution Running Head: Colonocyte keratins couple to nuclear lamina Corresponding author: Diana M. Toivola Cell Biology/Biosciences, Faculty of Science and Engineering, Åbo Akademi University Tykistökatu 6A, FIN-20520 Turku, Finland Telephone: +358 2 2154092 E-mail: [email protected] Keywords: Keratins, lamin, intermediate filament, colon epithelial cells, LINC proteins, proliferation, pRb, YAP bioRxiv preprint doi: https://doi.org/10.1101/2020.06.22.164467; this version posted June 22, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. -

Human Induced Pluripotent Stem Cell–Derived Podocytes Mature Into Vascularized Glomeruli Upon Experimental Transplantation
BASIC RESEARCH www.jasn.org Human Induced Pluripotent Stem Cell–Derived Podocytes Mature into Vascularized Glomeruli upon Experimental Transplantation † Sazia Sharmin,* Atsuhiro Taguchi,* Yusuke Kaku,* Yasuhiro Yoshimura,* Tomoko Ohmori,* ‡ † ‡ Tetsushi Sakuma, Masashi Mukoyama, Takashi Yamamoto, Hidetake Kurihara,§ and | Ryuichi Nishinakamura* *Department of Kidney Development, Institute of Molecular Embryology and Genetics, and †Department of Nephrology, Faculty of Life Sciences, Kumamoto University, Kumamoto, Japan; ‡Department of Mathematical and Life Sciences, Graduate School of Science, Hiroshima University, Hiroshima, Japan; §Division of Anatomy, Juntendo University School of Medicine, Tokyo, Japan; and |Japan Science and Technology Agency, CREST, Kumamoto, Japan ABSTRACT Glomerular podocytes express proteins, such as nephrin, that constitute the slit diaphragm, thereby contributing to the filtration process in the kidney. Glomerular development has been analyzed mainly in mice, whereas analysis of human kidney development has been minimal because of limited access to embryonic kidneys. We previously reported the induction of three-dimensional primordial glomeruli from human induced pluripotent stem (iPS) cells. Here, using transcription activator–like effector nuclease-mediated homologous recombination, we generated human iPS cell lines that express green fluorescent protein (GFP) in the NPHS1 locus, which encodes nephrin, and we show that GFP expression facilitated accurate visualization of nephrin-positive podocyte formation in -

Pflugers Final
CORE Metadata, citation and similar papers at core.ac.uk Provided by Serveur académique lausannois A comprehensive analysis of gene expression profiles in distal parts of the mouse renal tubule. Sylvain Pradervand2, Annie Mercier Zuber1, Gabriel Centeno1, Olivier Bonny1,3,4 and Dmitri Firsov1,4 1 - Department of Pharmacology and Toxicology, University of Lausanne, 1005 Lausanne, Switzerland 2 - DNA Array Facility, University of Lausanne, 1015 Lausanne, Switzerland 3 - Service of Nephrology, Lausanne University Hospital, 1005 Lausanne, Switzerland 4 – these two authors have equally contributed to the study to whom correspondence should be addressed: Dmitri FIRSOV Department of Pharmacology and Toxicology, University of Lausanne, 27 rue du Bugnon, 1005 Lausanne, Switzerland Phone: ++ 41-216925406 Fax: ++ 41-216925355 e-mail: [email protected] and Olivier BONNY Department of Pharmacology and Toxicology, University of Lausanne, 27 rue du Bugnon, 1005 Lausanne, Switzerland Phone: ++ 41-216925417 Fax: ++ 41-216925355 e-mail: [email protected] 1 Abstract The distal parts of the renal tubule play a critical role in maintaining homeostasis of extracellular fluids. In this review, we present an in-depth analysis of microarray-based gene expression profiles available for microdissected mouse distal nephron segments, i.e., the distal convoluted tubule (DCT) and the connecting tubule (CNT), and for the cortical portion of the collecting duct (CCD) (Zuber et al., 2009). Classification of expressed transcripts in 14 major functional gene categories demonstrated that all principal proteins involved in maintaining of salt and water balance are represented by highly abundant transcripts. However, a significant number of transcripts belonging, for instance, to categories of G protein-coupled receptors (GPCR) or serine-threonine kinases exhibit high expression levels but remain unassigned to a specific renal function. -

Typology and Profile of Spine Muscles. Structure of Myofibrils and Role of Protein Components – Review of Current Literature
Ovidius University Annals, Series Physical Education and Sport / SCIENCE, MOVEMENT AND HEALTH Vol. XI, ISSUE 2 Supplement, 2011, Romania The JOURNAL is nationally acknowledged by C.N.C.S.I.S., being included in the B+ category publications, 2008-2011. The journal is indexed in: Ebsco, SPORTDiscus, INDEX COPERNICUS JOURNAL MASTER LIST, DOAJ DIRECTORY OF OPEN ACCES JOURNALS, Caby, Gale Cengace Learning TYPOLOGY AND PROFILE OF SPINE MUSCLES. STRUCTURE OF MYOFIBRILS AND ROLE OF PROTEIN COMPONENTS – REVIEW OF CURRENT LITERATURE STRATON ALEXANDRU1, ENE-VOICULESCU CARMEN1, GIDU DIANA1, PETRESCU ANDREI1 Abstract. Muscular profile of spine muscles has a great importance in trunk stability. It seems that muscles which support the spine show a high content of red muscle fiber with a cross section area equal or higher than white muscle fibers. It is possible that lumbar extensor muscles to have different functional capacity between sexes. Most of the myofibril structural proteins except protein actin and protein myosin have a role in maintaining the structural integrity of muscle cell. Key words: muscle, fibres, myofibrils, proteins, spine. thoracic muscle structure lying superficial and deep is Introduction composed of 74% type I fibers, lumbar muscle Proper understanding of muscle fibers profile of structure located superficially is composed of 57% muscles supporting the spine and the role of muscle type I fibers and lumbar muscle structure located deep cell structural proteins leads to better achievements in is composed of 63% type I fibers. Type I muscle fiber performance training and rehabilitation. diameter is significantly larger than that of type II fibers. Another study, conducted on 42 patients Muscle fibers typology divided into two groups - 21 patients with lumbar Skeletal muscle contains two major types of back pain and 21 patients without lumbar back pain muscle fibers: slow twitch red or type I fibers) and almost identical groups as gender, age and body mass fast twitch white or type II fibers).