The Phylogeny of 48 Alleles, Experimentally Verified at 21 Kb
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Other Blood Group Systems—Diego,Yt, Xg, Scianna, Dombrock
Review: other blood group systems—Diego, Yt, Xg, Scianna, Dombrock, Colton, Landsteiner- Wiener, and Indian K.M. B YRNE AND P.C. B YRNE Introduction Diego Blood Group System This review was prepared to provide a basic The Diego blood group system (ISBT: DI/010) has overview of “Other Blood Groups.” Some of the more expanded from its humble beginnings to now include major blood group systems, i.e., ABO, Rh, Kell, Duffy, up to 21 discrete antigens (Table 1). 3 Band 3, an anion and Kidd, are also reviewed in this issue and are not exchange, multi-pass membrane glycoprotein, is the covered here. The sheer mass of data on the MNS basic structure that carries the Diego system antigenic blood group system is so extensive and complicated determinants. 4 The gene that encodes the Band 3 that it justifies a review all of its own, and it is therefore protein is named SLC4A1 and its chromosomal location not discussed in this article. However, various aspects is 17q12–q21. 4 of MNS were described in recent papers in Di a and Di b are antithetical, resulting from a single Immunohematology. 1,2 nucleotide substitution (2561T>C) that gives rise to The blood group systems that are covered are those amino acid changes in the Band 3 protein (Leu854Pro). that most workers believe to have some degree of To date, the Di(a–b–) phenotype has not been clinical importance or interesting features: Diego (DI), described. The Di a and Di b antigens are resistant to Yt (YT), Xg (XG), Scianna (SC), Dombrock (DO), Colton treatment with the following enzymes/chemicals: (CO), Landsteiner-Wiener (LW), and Indian (IN). -

Genes and Human History
Genes and human history Gil McVean, Department of Statistics, Oxford Contact: [email protected] 2 3 • Where does the variation come from? • How old are the genetic differences between us? • Are these differences important? How different are our genomes? 5 Serological techniques for detecting variation Rabbit Anti-A antibodies Human A A B AB O 6 Blood group systems in humans • 28 known systems – 39 genes, 643 alleles System Genes Alleles Knops CR1 24+ ABO ABO 102 Landsteiner- ICAM4 3 Wiener Colton C4A, C4B 7+ Lewis FUT3, FUT6 14/20 Chido-rodgers AQP1 7 Lutheran LU 16 Colton DAF 10 MNS GYPA,GYPB,G 43 Diego SLC4A1 78 YPE Dombrock DO 9 OK BSG 2 Duffy FY 9 P-related A4GALT, 14/5 Gerbich GYPC 9 B3GALT3 GIL AQP3 2 RAPH-MER2 CD151 3 H/h FUT1, FUT2 27/22 Rh RHCE, RHD, 129 I GCNT2 7 RHAG Indian CD44 2 Scianna ERMAP 4 Kell KEL, XK 33/30 Xg XG, CD99 - Kidd SLC14A1 8 YT ACHE 4 http://www.bioc.aecom.yu.edu/bgmut/summary.htm 7 Protein electroporesis • Changes in mass/charge ratio resulting from amino acid substitutions in proteins can be detected Starch or agar gel -- +- + +- - - - -- - + - + +- -- Direction of travel • In humans, about 30% of all loci show polymorphism with a 6% chance of a pair of randomly drawn alleles at a locus being different Lewontin and Hubby (1966) Harris(1966) 8 The rise of DNA sequencing GATAAGACGGTGATACTCACGCGACGGGCTTGGGCGCCGACTCGTTCAGACGGTGACCCAACTTATCCGATCGACCC CGGGTCCCGATTTAGACTCGGTATCATTTCTGGTGATTATCGCCTGCAGGTTCAAGAACACGTTTGCAGCAAGAAGT GAGGGATTTTGTCAGTGATCCCAGTCTACGGAGCCAGTCACCTCTGGTAGTGAAATTTTATTCGTTCATCTTCATAT AAGTCGCAGACCGCACGATGGGGGACAGAATACTCGCACAGGAAGAACCGCGATGAACCGAGGTAACCTAACATCCT -
Primepcr™Assay Validation Report
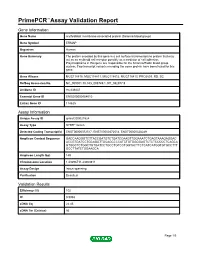
PrimePCR™Assay Validation Report Gene Information Gene Name erythroblast membrane-associated protein (Scianna blood group) Gene Symbol ERMAP Organism Human Gene Summary The protein encoded by this gene is a cell surface transmembrane protein that may act as an erythroid cell receptor possibly as a mediator of cell adhesion. Polymorphisms in this gene are responsible for the Scianna/Radin blood group system. Two transcript variants encoding the same protein have been found for this gene. Gene Aliases MGC118810, MGC118811, MGC118812, MGC118813, PRO2801, RD, SC RefSeq Accession No. NC_000001.10, NG_008749.1, NT_032977.9 UniGene ID Hs.439437 Ensembl Gene ID ENSG00000164010 Entrez Gene ID 114625 Assay Information Unique Assay ID qHsaCID0021524 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000372517, ENST00000372514, ENST00000328249 Amplicon Context Sequence GACCAAGGGTCTTACCGATGTCTGATCCAAGTTGGAAATCTGAGTAAAGAGGAC ACCGTGATCCTGCAGGTTGCAGCCCCATCTGTGGGGAGTCTCTCCCCCTCAGCA GTGGCTCTGGCTGTGATCCTGCCTGTCCTGGTACTTCTCATCATGGTGTGCCTTT GCCTTATCTGGAAGCA Amplicon Length (bp) 149 Chromosome Location 1:43296711-43300811 Assay Design Intron-spanning Purification Desalted Validation Results Efficiency (%) 102 R2 0.9968 cDNA Cq 22.45 cDNA Tm (Celsius) 86 Page 1/5 PrimePCR™Assay Validation Report gDNA Cq 40.23 Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 2/5 PrimePCR™Assay Validation Report ERMAP, Human Amplification Plot Amplification of cDNA generated from 25 ng of universal reference -

ERMAP Antibody (Monoclonal) (M01) Mouse Monoclonal Antibody Raised Against a Partial Recombinant ERMAP
10320 Camino Santa Fe, Suite G San Diego, CA 92121 Tel: 858.875.1900 Fax: 858.622.0609 ERMAP Antibody (monoclonal) (M01) Mouse monoclonal antibody raised against a partial recombinant ERMAP. Catalog # AT1943a Specification ERMAP Antibody (monoclonal) (M01) - Product Information Application WB, E Primary Accession Q96PL5 Other Accession NM_001017922 Reactivity Human Host mouse Clonality Monoclonal Isotype IgG2a Kappa Calculated MW 52605 ERMAP Antibody (monoclonal) (M01) - Additional Information Antibody Reactive Against Recombinant Protein.Western Blot detection against Gene ID 114625 Immunogen (36.74 KDa) . Other Names Erythroid membrane-associated protein, hERMAP, Radin blood group antigen, Scianna blood group antigen, ERMAP, RD, SC Target/Specificity ERMAP (NP_001017922, 376 a.a. ~ 475 a.a) partial recombinant protein with GST tag. MW of the GST tag alone is 26 KDa. Dilution WB~~1:500~1000 Format ERMAP monoclonal antibody (M01), clone Clear, colorless solution in phosphate 6F8 Western Blot analysis of ERMAP buffered saline, pH 7.2 . expression in HeLa ( (Cat # AT1943a ) Storage Store at -20°C or lower. Aliquot to avoid repeated freezing and thawing. Precautions ERMAP Antibody (monoclonal) (M01) is for research use only and not for use in diagnostic or therapeutic procedures. ERMAP Antibody (monoclonal) (M01) - Detection limit for recombinant GST tagged Page 1/2 10320 Camino Santa Fe, Suite G San Diego, CA 92121 Tel: 858.875.1900 Fax: 858.622.0609 Protocols ERMAP is approximately 0.1ng/ml as a capture antibody. Provided below are standard protocols that you may find useful for product applications. ERMAP Antibody (monoclonal) (M01) - • Western Blot Background • Blocking Peptides • Dot Blot The protein encoded by this gene is a cell • Immunohistochemistry surface transmembrane protein that may act • Immunofluorescence as an erythroid cell receptor, possibly as a • Immunoprecipitation mediator of cell adhesion. -

Mirepoix LLC on Behalf of Caris Life Sciences, June 22, 2020 No Revisions to These Recommendations
0211U Oncology (pan-tumor), DNA and RNA by next generation sequencing, utilizing formalin-fixed paraffin-embedded tissue, interpretative report for single nucleotide variants, copy number alterations, tumor mutational burden, and microsatellite instability, with therapy association Submitted by Joel de Jesus, Mirepoix LLC on behalf of Caris Life Sciences, June 22, 2020 No revisions to these recommendations 0211U: MI Cancer Seek™ - NGS analysis C. Public Comment Rationale • Recommendation to crosswalk 0211U MI Cancer Seek is a combined whole transcriptome AND whole exome to 0019U (NLA=$3675.00) + 0036U sequencing test that is equivalent to (NLA=$4780.00) performing both: • 0019U: Oncology, RNA, gene expression • 1x multiplier for each by whole transcriptome sequencing, formalin-fixed paraffin-embedded tissue • Recommendation of an NLA equal to or fresh frozen tissue, predictive $8455 for 0211U algorithm reported as potential targets for therapeutic agents • 0036U: Exome (ie, somatic mutations), paired formalin-fixed paraffin-embedded tumor tissue and normal specimen, sequence analyses Submitted by Joel de Jesus, Mirepoix LLC on behalf of Caris Life Sciences, June 22, 2020 No revisions to these recommendations 0211U: MI Cancer Seek™ - NGS analysis C. 0019U* 0211U 0036U** Assay type Whole Whole Whole Whole Transcriptome Transcriptome Exome Exome Sequencing Sequencing Sequencing Sequencing Platform NGS NGS (Illumina) NGS (Illumina) RNASeq RNASeq & DNASeq DNASeq Samples tested FFPE & frozen tumor tissue FFPE tumor tissue FFPE tumor tissue -

Blood Product Molecular Antigen Typing
UnitedHealthcare® Medicare Advantage Policy Guideline Blood Product Molecular Antigen Typing Guideline Number: MPG388.01 Approval Date: February 10, 2021 Terms and Conditions Table of Contents Page Related Medicare Advantage Policy Guidelines Policy Summary ............................................................................. 1 • Clinical Diagnostic Laboratory Services Applicable Codes .......................................................................... 2 • Molecular Pathology/Molecular References ..................................................................................... 7 Diagnostics/Genetic Testing Guideline History/Revision Information ....................................... 8 Purpose .......................................................................................... 9 Related Medicare Advantage Coverage Summaries Terms and Conditions ................................................................... 9 • Genetic Testing • Laboratory Tests and Services Policy Summary See Purpose Overview Based on the Centers for Medicare & Medicaid Services (CMS) Program Integrity Manual (100-08), this policy addresses the circumstances under which the item or service is reasonable and necessary under the Social Security Act, §1862(a)(1)(A). For laboratory services, a service can be reasonable and necessary if the service is safe and effective; and appropriate, including the duration and frequency that is considered appropriate for the item or service, in terms of whether it is furnished in accordance with accepted -

An Introduction to Blood Groups
1405153490_4_001.qxd 8/16/06 9:21 AM Page 1 An introduction to blood groups CHAPTER 1 What is a blood group? In 1900, Landsteiner showed that people could be divided into three groups (now called A, B, and O) on the basis of whether their red cells clumped when mixed with separated sera from people. A fourth group (AB) was soon found. This is the origin of the term ‘blood group’. A blood group could be defined as, ‘An inherited character of the red cell surface, detected by a specific alloantibody’. Do blood groups have to be present on red cells? This is the usual meaning, though platelet- and neutrophil-specific antigens might also be called blood groups. In this book only red cell surface antigens are considered. Blood groups do not have to be red cell specific, or even blood cell specific, and most are also detected on other cell types. Blood groups do have to be detected by a specific antibody: polymorphisms suspected of being present on the red cell surface, but only detected by other means, such as DNA sequencing, are not blood groups. Furthermore, the antibodies must be alloantibod- ies, implying that some individuals lack the blood group. Blood group antigens may be: • proteins; • glycoproteins, with the antibody recognising primarily the polypeptide backbone; • glycoproteins, with the antibody recognising the carbohydrate moiety; • glycolipids, with the antibody recognising the carbohydrate portion. Blood group polymorphisms may be as fundamental as representing the presence or absence of the whole macromolecule (e.g. RhD), or as minor as a single amino acid change (e.g. -

Characterisation of Weak and Null Phenotypes in the KEL and JK Blood Group Systems
Characterisation of weak and null phenotypes in the KEL and JK blood group systems Sjöberg Wester, Elisabet 2010 Link to publication Citation for published version (APA): Sjöberg Wester, E. (2010). Characterisation of weak and null phenotypes in the KEL and JK blood group systems. Department of Laboratory Medicine, Lund University. Total number of authors: 1 General rights Unless other specific re-use rights are stated the following general rights apply: Copyright and moral rights for the publications made accessible in the public portal are retained by the authors and/or other copyright owners and it is a condition of accessing publications that users recognise and abide by the legal requirements associated with these rights. • Users may download and print one copy of any publication from the public portal for the purpose of private study or research. • You may not further distribute the material or use it for any profit-making activity or commercial gain • You may freely distribute the URL identifying the publication in the public portal Read more about Creative commons licenses: https://creativecommons.org/licenses/ Take down policy If you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediately and investigate your claim. LUND UNIVERSITY PO Box 117 221 00 Lund +46 46-222 00 00 Download date: 06. Oct. 2021 Elisabet Sjöberg Wester Elisabet Sjöberg Wester Characterisation of weak and null phenotypes in the KEL and JK blood group systems Characterisation of weak -

Blood Group Genotyping Genotipagem De Grupos Sangüíneos
Castilho L et al Rev. bras. hematol. hemoter. 2004;26(2):135-140 Revisão / Review Blood group genotyping Genotipagem de grupos sangüíneos Lilian Castilho1 Accurate phenotyping of red blood cells (RBCs) can be difficult in transfusion- Jordão Pellegrino Júnior2 dependent patients such as those with thalassemia and sickle cells anemia because of the presence of previously transfused RBCs in the patient’s circulation. Recently, the molecular basis associated with the expression of many blood group antigens was established. This allowed the development of a plethora of polymerase chain reaction-based tests for identification of the blood group antigens by testing DNA. The determination of blood group polymorphism at the genomic level facilitates the resolution of clinical problems that cannot be addressed by hemagglutination. They are useful to (a) determine antigen types for which currently available antibodies are weakly reactive; (b) type patients who have been recently transfused; (c) identify fetuses at risk for hemolytic disease of the newborn; and (d) to increase the reliability of repositories of antigen negative RBCs for transfusion. It is important to note that PCR based assays are prone to different types of errors that those observed with hemagglutination assays. For instance, contamination with amplified products may lead to false positive test results. In addition, the identification of a particular genotype does not necessarily mean that the antigen will be expressed on the RBC membrane. Rev. bras. hematol. hemoter. 2004;26(2):135-140. Key words: Blood group antigens; DNA technology; transfused patients; hemagglutination; maternal-fetal medicine. Introduction chronically transfused patients. Alloimmunization leads to an increased risk of transfusion reactions, reducing the Blood group antigens are polymorphisms of proteins available pool of compatible blood for transfusion in and carbohydrates on the outside surface of the red blood subsequent crises. -

Seltene Bluttypen Und Antikörper Gegen Hochfrequente Erythrozytenantigene
Seltene Bluttypen und Antikörper gegen hochfrequente Erythrozytenantigene Christof Jungbauer, ÖGBT 2013 Austrian Red Cross, Blood Service Content 1 Introduction 2 Antibody specificities to high frequency antigens: incidence in Europe 3 Impact of migration 4 Supply of rare blood Austrian Red Cross, Blood Service 2 Patients with red cell antibodies Prevalence of the compatible blood type Two situations may challenge an adequate blood supply: • Multiple antibody specificities e.g., anti-e, -Fy a, -Jk a (compatible blood: 1:670) • Specificity to a high frequency antigen (HFA) e.g., anti-k (compatible blood: 1:500) e.g., anti-Kp b (compatible blood: 1:10.000) Austrian Red Cross, Blood Service 3 Patients with red cell antibodies Prevalence of the compatible blood type Two situations may challenge an adequate blood supply: • Multiple antibody specificities e.g., anti-e, -Fy a, -Jk a (compatible blood: 1:670) • Specificity to a high frequency antigen (HFA) e.g., anti-U Blacks 1: 50 Caucasians 1: >10.000 Austrian Red Cross, Blood Service 4 Patients with red cell antibodies Prevalence of the compatible blood type Some (rare) blood types have an unequal prevalence in different ethnic populations While blood establishments have some preparedness for the “local” specificities to HFA, .... reagents and compatible donors for the supply of “exotic” rare blood types are missing Austrian Red Cross, Blood Service 5 modified from: http://www.isbtweb.org/fileadmin/user_upload/WP_on_Red_Cell_Immunogenetics_and/ ISBT BLOOD GROUP SYSTEMS Table_of_blood_group_antigens_within_systems_v2.0_110914.pdf -

Widespread Balancing Selection and Pathogen-Driven Selection at Blood Group Antigen Genes
Downloaded from genome.cshlp.org on October 2, 2021 - Published by Cold Spring Harbor Laboratory Press Letter Widespread balancing selection and pathogen-driven selection at blood group antigen genes Matteo Fumagalli,1,2 Rachele Cagliani,1 Uberto Pozzoli,1 Stefania Riva,1 Giacomo P. Comi,3 Giorgia Menozzi,1 Nereo Bresolin,1,3 and Manuela Sironi1,4 1Scientific Institute IRCCS E. Medea, Bioinformatic Lab, 23842 Bosisio Parini (LC), Italy; 2Bioengineering Department, Politecnico di Milano, 20133 Milan, Italy; 3Dino Ferrari Centre, Department of Neurological Sciences, University of Milan, IRCCS Ospedale Maggiore Policlinico, Mangiagalli and Regina Elena Foundation, 20100 Milan, Italy Historically, allelic variations in blood group antigen (BGA) genes have been regarded as possible susceptibility factors for infectious diseases. Since host–pathogen interactions are major determinants in evolution, BGAs can be thought of as selection targets. In order to verify this hypothesis, we obtained an estimate of pathogen richness for geographic locations corresponding to 52 populations distributed worldwide; after correction for multiple tests and for variables different from selective forces, significant correlations with pathogen richness were obtained for multiple variants at 11 BGA loci out of 26. In line with this finding, we demonstrate that three BGA genes, namely CD55, CD151, and SLC14A1, have been subjected to balancing selection, a process, rare outside MHC genes, which maintains variability at a locus. Moreover, we identified a gene region immediately upstream the transcription start site of FUT2 which has undergone non-neutral evolution independently from the coding region. Finally, in the case of BSG, we describe the presence of a highly divergent hap- lotype clade and the possible reasons for its maintenance, including frequency-dependent balancing selection, are dis- cussed. -

Blood Group Genetics
Blood Group Genetics Department of Medical Genetics Medical University of Warsaw © Zakład Genetyki Medycznej 2013 Classical genetics Classical genetics laws: • Mendel's First Law – Law of Segregation • Mendel's Second Law – Law of Independent Assortment • Co-dominance – phenotypic expression of gene’s alleles is simultaneous and independent • Epistasis – phenomenon in which one gene influences the phenotypic expression of another gene, while not being its allele Blood Group Systems Blood Group – antigenic Group system – consists of one or more determinants on the antigens controlled at a single gene locus, or by two or more very closely surface of erythrocytes linked homologous genes distinguished by the Collection – consist of serologically, immune system in biochemically, or genetically related individuals without a antigens, which do not fit the criteria particular antigen required for system status (200 series) (alloantibodies) 901 Series – (or ’public’) contains antigens with an incidence of greater than 90% and which cannot be included in a system or collectioni 700 Series – (or ’private’) contains • 285 antigens antigens with an incidence of less than 1% and which cannot be included in a • 33 group systems system or collection Red blood cells group systems Nr Nazwa Symbol Gen Chromosom Nr Nazwa Symbol Gen Chromosom 001 ABO ABO ABO 9q34.2 018 H H FUT1 19q13.33 GYPA, GYPB, 002 MNS MNS GYPE 4q31.21 019 Kx XK XK Xp21.1 003 P1PK P1PK A4GALT 22q13.2 020 Gerbich GE GYPC 2q14.3 004 Rh RH RHD, RHCE 1p36.11 021 Cromer CROM CD55 1q32.2 005