Next Generation Sequencing-Based Genotyping of Human Blood Groups: FY, JK and ABO Genes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Other Blood Group Systems—Diego,Yt, Xg, Scianna, Dombrock
Review: other blood group systems—Diego, Yt, Xg, Scianna, Dombrock, Colton, Landsteiner- Wiener, and Indian K.M. B YRNE AND P.C. B YRNE Introduction Diego Blood Group System This review was prepared to provide a basic The Diego blood group system (ISBT: DI/010) has overview of “Other Blood Groups.” Some of the more expanded from its humble beginnings to now include major blood group systems, i.e., ABO, Rh, Kell, Duffy, up to 21 discrete antigens (Table 1). 3 Band 3, an anion and Kidd, are also reviewed in this issue and are not exchange, multi-pass membrane glycoprotein, is the covered here. The sheer mass of data on the MNS basic structure that carries the Diego system antigenic blood group system is so extensive and complicated determinants. 4 The gene that encodes the Band 3 that it justifies a review all of its own, and it is therefore protein is named SLC4A1 and its chromosomal location not discussed in this article. However, various aspects is 17q12–q21. 4 of MNS were described in recent papers in Di a and Di b are antithetical, resulting from a single Immunohematology. 1,2 nucleotide substitution (2561T>C) that gives rise to The blood group systems that are covered are those amino acid changes in the Band 3 protein (Leu854Pro). that most workers believe to have some degree of To date, the Di(a–b–) phenotype has not been clinical importance or interesting features: Diego (DI), described. The Di a and Di b antigens are resistant to Yt (YT), Xg (XG), Scianna (SC), Dombrock (DO), Colton treatment with the following enzymes/chemicals: (CO), Landsteiner-Wiener (LW), and Indian (IN). -

Genetic Screening for the Vel- Phenotype Circumvents Difficult Serological Screening Due to Variable Vel Expression Levels
UvA-DARE (Digital Academic Repository) Genetic basis of rare blood group variants Wigman, L. Publication date 2013 Link to publication Citation for published version (APA): Wigman, L. (2013). Genetic basis of rare blood group variants. General rights It is not permitted to download or to forward/distribute the text or part of it without the consent of the author(s) and/or copyright holder(s), other than for strictly personal, individual use, unless the work is under an open content license (like Creative Commons). Disclaimer/Complaints regulations If you believe that digital publication of certain material infringes any of your rights or (privacy) interests, please let the Library know, stating your reasons. In case of a legitimate complaint, the Library will make the material inaccessible and/or remove it from the website. Please Ask the Library: https://uba.uva.nl/en/contact, or a letter to: Library of the University of Amsterdam, Secretariat, Singel 425, 1012 WP Amsterdam, The Netherlands. You will be contacted as soon as possible. UvA-DARE is a service provided by the library of the University of Amsterdam (https://dare.uva.nl) Download date:28 Sep 2021 Chapter 6 Genetic screening for the Vel- phenotype circumvents difficult serological screening due to variable Vel expression levels Lonneke Haer-Wigman1 Shabnam Solati1 Aïcha Ait Soussan1 Erik Beckers2 Pim van der Harst3 Marga van Hulst-Sundermeijer4 Peter Ligthart1 Dick van Rhenen5 Hein Schepers3 Tamara Stegmann1 Masja de Haas1 C. Ellen van der Schoot1 1 Sanquin Research, Amsterdam and Landsteiner Laboratory, Academic Medical Centre, University of Amsterdam, Amsterdam, the Netherlands 2 Maastricht University Medical Centre+, Maastricht, The Netherlands 3 University Medical Center Groningen, University of Groningen, Groningen, The Netherlands 4 Sanquin Diagnostic Services, Amsterdam, The Netherlands 5 Sanquin Blood Bank, Rotterdam, Netherlands Manuscript under preparation Chapter 6 Abstract Background: Serological determination of the Vel- phenotype is challenging due to variable Vel expression levels. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Journal of Blood Group Serology and Molecular Genetics Volume 34, Number 1, 2018 CONTENTS
Journal of Blood Group Serology and Molecular Genetics VOLUME 34, N UMBER 1, 2018 This issue of Immunohematology is supported by a contribution from Grifols Diagnostics Solutions, Inc. Dedicated to advancement and education in molecular and serologic immunohematology Immunohematology Journal of Blood Group Serology and Molecular Genetics Volume 34, Number 1, 2018 CONTENTS S EROLOGIC M ETHOD R EVIEW 1 Warm autoadsorption using ZZAP F.M. Tsimba-Chitsva, A. Caballero, and B. Svatora R EVIEW 4 Proceedings from the International Society of Blood Transfusion Working Party on Immunohaematology Workshop on the Clinical Significance of Red Blood Cell Alloantibodies, Friday, September 2, 2016, Dubai A brief overview of clinical significance of blood group antibodies M.J. Gandhi, D.M. Strong, B.I. Whitaker, and E. Petrisli C A S E R EPORT 7 Management of pregnancy sensitized with anti-Inb with monocyte monolayer assay and maternal blood donation R. Shree, K.K. Ma, L.S. Er and M. Delaney R EVIEW 11 Proceedings from the International Society of Blood Transfusion Working Party on Immunohaematology Workshop on the Clinical Significance of Red Blood Cell Alloantibodies, Friday, September 2, 2016, Dubai A review of in vitro methods to predict the clinical significance of red blood cell alloantibodies S.J. Nance S EROLOGIC M ETHOD R EVIEW 16 Recovery of autologous sickle cells by hypotonic wash E. Wilson, K. Kezeor, and M. Crosby TO THE E DITOR 19 The devil is in the details: retention of recipient group A type 5 years after a successful allogeneic bone marrow transplant from a group O donor L.L.W. -

Genes and Human History
Genes and human history Gil McVean, Department of Statistics, Oxford Contact: [email protected] 2 3 • Where does the variation come from? • How old are the genetic differences between us? • Are these differences important? How different are our genomes? 5 Serological techniques for detecting variation Rabbit Anti-A antibodies Human A A B AB O 6 Blood group systems in humans • 28 known systems – 39 genes, 643 alleles System Genes Alleles Knops CR1 24+ ABO ABO 102 Landsteiner- ICAM4 3 Wiener Colton C4A, C4B 7+ Lewis FUT3, FUT6 14/20 Chido-rodgers AQP1 7 Lutheran LU 16 Colton DAF 10 MNS GYPA,GYPB,G 43 Diego SLC4A1 78 YPE Dombrock DO 9 OK BSG 2 Duffy FY 9 P-related A4GALT, 14/5 Gerbich GYPC 9 B3GALT3 GIL AQP3 2 RAPH-MER2 CD151 3 H/h FUT1, FUT2 27/22 Rh RHCE, RHD, 129 I GCNT2 7 RHAG Indian CD44 2 Scianna ERMAP 4 Kell KEL, XK 33/30 Xg XG, CD99 - Kidd SLC14A1 8 YT ACHE 4 http://www.bioc.aecom.yu.edu/bgmut/summary.htm 7 Protein electroporesis • Changes in mass/charge ratio resulting from amino acid substitutions in proteins can be detected Starch or agar gel -- +- + +- - - - -- - + - + +- -- Direction of travel • In humans, about 30% of all loci show polymorphism with a 6% chance of a pair of randomly drawn alleles at a locus being different Lewontin and Hubby (1966) Harris(1966) 8 The rise of DNA sequencing GATAAGACGGTGATACTCACGCGACGGGCTTGGGCGCCGACTCGTTCAGACGGTGACCCAACTTATCCGATCGACCC CGGGTCCCGATTTAGACTCGGTATCATTTCTGGTGATTATCGCCTGCAGGTTCAAGAACACGTTTGCAGCAAGAAGT GAGGGATTTTGTCAGTGATCCCAGTCTACGGAGCCAGTCACCTCTGGTAGTGAAATTTTATTCGTTCATCTTCATAT AAGTCGCAGACCGCACGATGGGGGACAGAATACTCGCACAGGAAGAACCGCGATGAACCGAGGTAACCTAACATCCT -

Análise Integrativa De Perfis Transcricionais De Pacientes Com
UNIVERSIDADE DE SÃO PAULO FACULDADE DE MEDICINA DE RIBEIRÃO PRETO PROGRAMA DE PÓS-GRADUAÇÃO EM GENÉTICA ADRIANE FEIJÓ EVANGELISTA Análise integrativa de perfis transcricionais de pacientes com diabetes mellitus tipo 1, tipo 2 e gestacional, comparando-os com manifestações demográficas, clínicas, laboratoriais, fisiopatológicas e terapêuticas Ribeirão Preto – 2012 ADRIANE FEIJÓ EVANGELISTA Análise integrativa de perfis transcricionais de pacientes com diabetes mellitus tipo 1, tipo 2 e gestacional, comparando-os com manifestações demográficas, clínicas, laboratoriais, fisiopatológicas e terapêuticas Tese apresentada à Faculdade de Medicina de Ribeirão Preto da Universidade de São Paulo para obtenção do título de Doutor em Ciências. Área de Concentração: Genética Orientador: Prof. Dr. Eduardo Antonio Donadi Co-orientador: Prof. Dr. Geraldo A. S. Passos Ribeirão Preto – 2012 AUTORIZO A REPRODUÇÃO E DIVULGAÇÃO TOTAL OU PARCIAL DESTE TRABALHO, POR QUALQUER MEIO CONVENCIONAL OU ELETRÔNICO, PARA FINS DE ESTUDO E PESQUISA, DESDE QUE CITADA A FONTE. FICHA CATALOGRÁFICA Evangelista, Adriane Feijó Análise integrativa de perfis transcricionais de pacientes com diabetes mellitus tipo 1, tipo 2 e gestacional, comparando-os com manifestações demográficas, clínicas, laboratoriais, fisiopatológicas e terapêuticas. Ribeirão Preto, 2012 192p. Tese de Doutorado apresentada à Faculdade de Medicina de Ribeirão Preto da Universidade de São Paulo. Área de Concentração: Genética. Orientador: Donadi, Eduardo Antonio Co-orientador: Passos, Geraldo A. 1. Expressão gênica – microarrays 2. Análise bioinformática por module maps 3. Diabetes mellitus tipo 1 4. Diabetes mellitus tipo 2 5. Diabetes mellitus gestacional FOLHA DE APROVAÇÃO ADRIANE FEIJÓ EVANGELISTA Análise integrativa de perfis transcricionais de pacientes com diabetes mellitus tipo 1, tipo 2 e gestacional, comparando-os com manifestações demográficas, clínicas, laboratoriais, fisiopatológicas e terapêuticas. -

In Vitro Selection for Adhesion of Plasmodium Falciparum-Infected Erythrocytes to ABO Antigens Does Not Affect Pfemp1 and RIFIN
www.nature.com/scientificreports OPEN In vitro selection for adhesion of Plasmodium falciparum‑infected erythrocytes to ABO antigens does not afect PfEMP1 and RIFIN expression William van der Puije1,2, Christian W. Wang 4, Srinidhi Sudharson 2, Casper Hempel 2, Rebecca W. Olsen 4, Nanna Dalgaard 4, Michael F. Ofori 1, Lars Hviid 3,4, Jørgen A. L. Kurtzhals 2,4 & Trine Staalsoe 2,4* Plasmodium falciparum causes the most severe form of malaria in humans. The adhesion of the infected erythrocytes (IEs) to endothelial receptors (sequestration) and to uninfected erythrocytes (rosetting) are considered major elements in the pathogenesis of the disease. Both sequestration and rosetting appear to involve particular members of several IE variant surface antigens (VSAs) as ligands, interacting with multiple vascular host receptors, including the ABO blood group antigens. In this study, we subjected genetically distinct P. falciparum parasites to in vitro selection for increased IE adhesion to ABO antigens in the absence of potentially confounding receptors. The selection resulted in IEs that adhered stronger to pure ABO antigens, to erythrocytes, and to various human cell lines than their unselected counterparts. However, selection did not result in marked qualitative changes in transcript levels of the genes encoding the best-described VSA families, PfEMP1 and RIFIN. Rather, overall transcription of both gene families tended to decline following selection. Furthermore, selection-induced increases in the adhesion to ABO occurred in the absence of marked changes in immune IgG recognition of IE surface antigens, generally assumed to target mainly VSAs. Our study sheds new light on our understanding of the processes and molecules involved in IE sequestration and rosetting. -

Journal of Blood Group Serology and Molecular Genetics Volume 33, Number 3, 2017 CONTENTS
Journal of Blood Group Serology and Molecular Genetics VOLUME 33, N UMBER 3, 2017 This issue of Immunohematology is supported by a contribution from Grifols Diagnostics Solutions, Inc. Dedicated to advancement and education in molecular and serologic immunohematology Immunohematology Journal of Blood Group Serology and Molecular Genetics Volume 33, Number 3, 2017 CONTENTS C ASE R EPO R T 99 ABO serology in a case of persistent weak A in a recipient following a group O–matched unrelated bone marrow transplant D.E. Grey, E.A. Fong, C. Cole, J. Jensen, and J. Finlayson O R IGINAL R EPO R T 105 Stability guidelines for dithiothreitol-treated red blood cell reagents used for antibody detection methods in patients treated with daratumumab W.L. Disbro C ASE R EPO R T 110 A LU:−16 individual with antibodies C. Éthier, C. Parent, A.-S. Lemay, N. Baillargeon, G. Laflamme, J. Lavoie, J. Perreault, and M. St-Louis C ASE R EPO R T 114 Postpartum acute hemolytic transfusion reactions associated with anti-Lea in two pregnancies complicated by preeclampsia M. Marchese O R IGINAL R EPO R T 119 Red blood cell phenotype prevalence in blood donors who self- identify as Hispanic C.A. Sheppard, N.L. Bolen, B. Eades, G. Ochoa-Garay, and M.H. Yazer R EVIEW 125 DEL Phenotype D.H. Kwon, S.G. Sandler, and W.A. Flegel 133 138 142 144 A NNOUN C EMENTS A DVE R TISEMENTS I NST R U C TIONS S UBS cr IPTION FO R A UTHO R S I NFO R M AT I O N E DITO R - IN -C HIEF E DITO R IAL B OA R D Sandra Nance, MS, MT(ASCP)SBB Philadelphia, Pennsylvania Patricia Arndt, MT(ASCP)SBB Geralyn M. -

Blood Group Genomics
BLOOD GROUP GENOMICS Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry analysis of 36 blood group alleles among 396 Thai samples reveals region-specific variants Philaiphon Jongruamklang,1 Christoph Gassner,2 Stefan Meyer,2 Aksarakorn Kummasook,3 Marion Darlison,1 Chayanun Boonlum,4 Surin Chanta,5 Beat M. Frey,2 Martin L. Olsson,1,6* and Jill R. Storry 1,6* lood group antigen polymorphism shows great BACKGROUND: Blood group phenotype variation has variation in different world populations. The been attributed to potential resistance to pathogen reason for this is not completely understood; invasion. Variation was mapped in blood donors from however, it has been attributed to both Lampang (northern region) and Saraburi (central region), B Thailand, where malaria is endemic. The previously unknown blood group allele profiles were characterized ABBREVIATIONS: MALDI-TOF MS 5 matrix-assisted laser and the data were correlated with phenotypes. The high desorption/ionization time-of-flight mass spectrometry; PCR- incidence of the Vel-negative phenotype previously ASP 5 polymerase chain reaction with allele-specific reported in Thais was investigated. primers; SNP(s) 5 single nucleotide polymorphism(s). STUDY DESIGN AND METHODS: DNA from 396 From 1Hematology and Transfusion Medicine, Department of blood donors was analyzed by matrix-assisted laser Laboratory Medicine, Lund University, Lund, Sweden; desorption/ionization–time-of-flight mass spectrometry. 2Molecular Diagnostics & Research (MOC), Blood Transfusion Outliers were investigated by serology and DNA Service Zurich,€ Zurich-Schlieren,€ Switzerland; 3Department of sequencing. Allele discrimination assays for SMIM1 Medical Technology, School of Allied Health Sciences, rs1175550A/G and ACKR1 rs118062001C/T were University of Phayao, Phayao, Thailand; 4Transfusion Medicine, performed and correlated with antigen expression. -
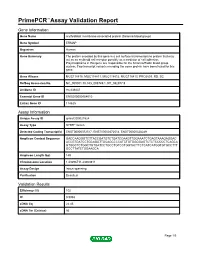
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name erythroblast membrane-associated protein (Scianna blood group) Gene Symbol ERMAP Organism Human Gene Summary The protein encoded by this gene is a cell surface transmembrane protein that may act as an erythroid cell receptor possibly as a mediator of cell adhesion. Polymorphisms in this gene are responsible for the Scianna/Radin blood group system. Two transcript variants encoding the same protein have been found for this gene. Gene Aliases MGC118810, MGC118811, MGC118812, MGC118813, PRO2801, RD, SC RefSeq Accession No. NC_000001.10, NG_008749.1, NT_032977.9 UniGene ID Hs.439437 Ensembl Gene ID ENSG00000164010 Entrez Gene ID 114625 Assay Information Unique Assay ID qHsaCID0021524 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000372517, ENST00000372514, ENST00000328249 Amplicon Context Sequence GACCAAGGGTCTTACCGATGTCTGATCCAAGTTGGAAATCTGAGTAAAGAGGAC ACCGTGATCCTGCAGGTTGCAGCCCCATCTGTGGGGAGTCTCTCCCCCTCAGCA GTGGCTCTGGCTGTGATCCTGCCTGTCCTGGTACTTCTCATCATGGTGTGCCTTT GCCTTATCTGGAAGCA Amplicon Length (bp) 149 Chromosome Location 1:43296711-43300811 Assay Design Intron-spanning Purification Desalted Validation Results Efficiency (%) 102 R2 0.9968 cDNA Cq 22.45 cDNA Tm (Celsius) 86 Page 1/5 PrimePCR™Assay Validation Report gDNA Cq 40.23 Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 2/5 PrimePCR™Assay Validation Report ERMAP, Human Amplification Plot Amplification of cDNA generated from 25 ng of universal reference -

ERMAP Antibody (Monoclonal) (M01) Mouse Monoclonal Antibody Raised Against a Partial Recombinant ERMAP
10320 Camino Santa Fe, Suite G San Diego, CA 92121 Tel: 858.875.1900 Fax: 858.622.0609 ERMAP Antibody (monoclonal) (M01) Mouse monoclonal antibody raised against a partial recombinant ERMAP. Catalog # AT1943a Specification ERMAP Antibody (monoclonal) (M01) - Product Information Application WB, E Primary Accession Q96PL5 Other Accession NM_001017922 Reactivity Human Host mouse Clonality Monoclonal Isotype IgG2a Kappa Calculated MW 52605 ERMAP Antibody (monoclonal) (M01) - Additional Information Antibody Reactive Against Recombinant Protein.Western Blot detection against Gene ID 114625 Immunogen (36.74 KDa) . Other Names Erythroid membrane-associated protein, hERMAP, Radin blood group antigen, Scianna blood group antigen, ERMAP, RD, SC Target/Specificity ERMAP (NP_001017922, 376 a.a. ~ 475 a.a) partial recombinant protein with GST tag. MW of the GST tag alone is 26 KDa. Dilution WB~~1:500~1000 Format ERMAP monoclonal antibody (M01), clone Clear, colorless solution in phosphate 6F8 Western Blot analysis of ERMAP buffered saline, pH 7.2 . expression in HeLa ( (Cat # AT1943a ) Storage Store at -20°C or lower. Aliquot to avoid repeated freezing and thawing. Precautions ERMAP Antibody (monoclonal) (M01) is for research use only and not for use in diagnostic or therapeutic procedures. ERMAP Antibody (monoclonal) (M01) - Detection limit for recombinant GST tagged Page 1/2 10320 Camino Santa Fe, Suite G San Diego, CA 92121 Tel: 858.875.1900 Fax: 858.622.0609 Protocols ERMAP is approximately 0.1ng/ml as a capture antibody. Provided below are standard protocols that you may find useful for product applications. ERMAP Antibody (monoclonal) (M01) - • Western Blot Background • Blocking Peptides • Dot Blot The protein encoded by this gene is a cell • Immunohistochemistry surface transmembrane protein that may act • Immunofluorescence as an erythroid cell receptor, possibly as a • Immunoprecipitation mediator of cell adhesion. -

Transfusion Medicine
Transfusion Medicine Dr. Raymond SM Wong Department of Medicine & Therapeutics Prince of Wales Hospital The Chinese University of Hong Kong Content Blood groups Cross-matching and pre-transfusion tests Blood components and blood products Complications of blood transfusion Blood transfusion in specific situations Blood groups ( 血型) Determined by the red cell antigens ( 紅血球抗原) About 400 red blood cell group antigens have been described Individual who lack a particular blood group antigen may produce antibodies ( 抗體) reacting with that antigen and may lead to a transfusion reaction ( 輸血 反應) ABO and rhesus ( 獼因子) groups are the most clinically significant blood groups Blood group antibodies Naturally occurring antibodies occur in plasma of subjects who lack the corresponding antigen and who have not been transfused or been pregnant Most important are anti-A and anti-B Immune antibodies Develop in response to exposure to antigens by transfusion or by trans-placental passage during pregnancy Most important is the Rhesus (Rh) antibody, anti-D ABO blood group system Consists of 3 allelic genes: A, B and O A and B gene control the synthesis of specific enzymes which transform the H substance Ceramide glu gal gnac gal fuc H antigen galnac Ceramide glu gal gnac gal A antigen fuc gal Ceramide glu gal gnac gal B antigen fuc Cell membrane ABO blood group system Phenotype Genotype Naturally occurring (表型) (基因型) Antigens antibodies O OO O Anti-A, anti-B A AA or AO A Anti-B B BB or BO B Anti-A AB AB AB None The O gene is an amorph (無效基因)