Identification, Characterization, and Utilization of Glycosyltransferases
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Federal Communications Commission Record FCC 94-206
9 FCC Red No. 18 Federal Communications Commission Record FCC 94-206 application if there are other compelling circumstances Before the that warrant approval. For the reasons set forth below, we Federal Communications Commission find that the proposed operation of WICD(TV) as a satellite Washington, D.C. 20554 is consistent with our policy. 3. In support of its request that WICD(TV) be permitted to operate as a satellite of WICS-TV, the applicant contends In Re Application of that its proposal meets the three criteria for presumptive grant. First, the applicant states, there is no overlap of the PLAINS TELEVISION PARTNERSHIP City Grade contours of the two stations. Second, (Assignor) WICD(TV)©s area of service is underserved in accordance with the Commission©s "transmission test." Finally, the applicant argues that no party would be willing and able to and File No. BALCT-931124KJ operate WICD(TV) on a stand-alone basis because of the nature of the Springfield-Decatur-Champaign market. In GUY GANNETT PUBLISHING CO. addition, the applicant argues that the Grade B overlap of (Assignee) WICS-TV and WICD(TV) only amounts to 2.6% of the population in the WICD(TV) contour and 2% in the For Consent to Assign the License for Station WICS-TV contour. The applicants point out that the Com WICD(TV), Champaign, IL mission found a similar overlap to be insubstantial when it approved the operation of WCCU(TV) (Urbana) as a sat ellite of WRSP-TV (Springfield). Springfield Independent MEMORANDUM OPINION AND ORDER Television, 3 FCC Red 1606, 1607 (1988). -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Who Pays Soundexchange: Q1 - Q3 2017
Payments received through 09/30/2017 Who Pays SoundExchange: Q1 - Q3 2017 Entity Name License Type ACTIVAIRE.COM BES AMBIANCERADIO.COM BES AURA MULTIMEDIA CORPORATION BES CLOUDCOVERMUSIC.COM BES COROHEALTH.COM BES CUSTOMCHANNELS.NET (BES) BES DMX MUSIC BES ELEVATEDMUSICSERVICES.COM BES GRAYV.COM BES INSTOREAUDIONETWORK.COM BES IT'S NEVER 2 LATE BES JUKEBOXY BES MANAGEDMEDIA.COM BES MEDIATRENDS.BIZ BES MIXHITS.COM BES MTI Digital Inc - MTIDIGITAL.BIZ BES MUSIC CHOICE BES MUSIC MAESTRO BES MUZAK.COM BES PRIVATE LABEL RADIO BES RFC MEDIA - BES BES RISE RADIO BES ROCKBOT, INC. BES SIRIUS XM RADIO, INC BES SOUND-MACHINE.COM BES STARTLE INTERNATIONAL INC. BES Stingray Business BES Stingray Music USA BES STORESTREAMS.COM BES STUDIOSTREAM.COM BES TARGET MEDIA CENTRAL INC BES Thales InFlyt Experience BES UMIXMEDIA.COM BES SIRIUS XM RADIO, INC CABSAT Stingray Music USA CABSAT MUSIC CHOICE PES MUZAK.COM PES SIRIUS XM RADIO, INC SDARS 181.FM Webcasting 3ABNRADIO (Christian Music) Webcasting 3ABNRADIO (Religious) Webcasting 8TRACKS.COM Webcasting 903 NETWORK RADIO Webcasting A-1 COMMUNICATIONS Webcasting ABERCROMBIE.COM Webcasting ABUNDANT RADIO Webcasting ACAVILLE.COM Webcasting *SoundExchange accepts and distributes payments without confirming eligibility or compliance under Sections 112 or 114 of the Copyright Act, and it does not waive the rights of artists or copyright owners that receive such payments. Payments received through 09/30/2017 ACCURADIO.COM Webcasting ACRN.COM Webcasting AD ASTRA RADIO Webcasting ADAMS RADIO GROUP Webcasting ADDICTEDTORADIO.COM Webcasting ADORATION Webcasting AGM BAKERSFIELD Webcasting AGM CALIFORNIA - SAN LUIS OBISPO Webcasting AGM NEVADA, LLC Webcasting AGM SANTA MARIA, L.P. -

Análise Integrativa De Perfis Transcricionais De Pacientes Com
UNIVERSIDADE DE SÃO PAULO FACULDADE DE MEDICINA DE RIBEIRÃO PRETO PROGRAMA DE PÓS-GRADUAÇÃO EM GENÉTICA ADRIANE FEIJÓ EVANGELISTA Análise integrativa de perfis transcricionais de pacientes com diabetes mellitus tipo 1, tipo 2 e gestacional, comparando-os com manifestações demográficas, clínicas, laboratoriais, fisiopatológicas e terapêuticas Ribeirão Preto – 2012 ADRIANE FEIJÓ EVANGELISTA Análise integrativa de perfis transcricionais de pacientes com diabetes mellitus tipo 1, tipo 2 e gestacional, comparando-os com manifestações demográficas, clínicas, laboratoriais, fisiopatológicas e terapêuticas Tese apresentada à Faculdade de Medicina de Ribeirão Preto da Universidade de São Paulo para obtenção do título de Doutor em Ciências. Área de Concentração: Genética Orientador: Prof. Dr. Eduardo Antonio Donadi Co-orientador: Prof. Dr. Geraldo A. S. Passos Ribeirão Preto – 2012 AUTORIZO A REPRODUÇÃO E DIVULGAÇÃO TOTAL OU PARCIAL DESTE TRABALHO, POR QUALQUER MEIO CONVENCIONAL OU ELETRÔNICO, PARA FINS DE ESTUDO E PESQUISA, DESDE QUE CITADA A FONTE. FICHA CATALOGRÁFICA Evangelista, Adriane Feijó Análise integrativa de perfis transcricionais de pacientes com diabetes mellitus tipo 1, tipo 2 e gestacional, comparando-os com manifestações demográficas, clínicas, laboratoriais, fisiopatológicas e terapêuticas. Ribeirão Preto, 2012 192p. Tese de Doutorado apresentada à Faculdade de Medicina de Ribeirão Preto da Universidade de São Paulo. Área de Concentração: Genética. Orientador: Donadi, Eduardo Antonio Co-orientador: Passos, Geraldo A. 1. Expressão gênica – microarrays 2. Análise bioinformática por module maps 3. Diabetes mellitus tipo 1 4. Diabetes mellitus tipo 2 5. Diabetes mellitus gestacional FOLHA DE APROVAÇÃO ADRIANE FEIJÓ EVANGELISTA Análise integrativa de perfis transcricionais de pacientes com diabetes mellitus tipo 1, tipo 2 e gestacional, comparando-os com manifestações demográficas, clínicas, laboratoriais, fisiopatológicas e terapêuticas. -

Tattler for Pdf 11/1
Volume XXIX • Number 5 • January 31, 2003 Grand Rapids Fall Book. Clear Channel’s Country WBCT is on top once again! WBCT 9.9-9.6, WSNX 8.1-6.8, WLAV 7.3-6.4, WOOD-FM THETHE 4.9-5.7, WOOD-AM 5.1-5.5, WLHT 4.6-5.2, WGRD 6.4-5.0, WKLQ 5.8- 4.7, WTRV 3.7-4.2, WBFX 3.8-4.0, WODJ 3.9-3.6, WJQK 2.5-2.8, WVTI MAIN STREET 2.8-2.3, WFGR 1.6-2.2, WBBL-AM 1.7-2.1, WMUS 1.5-1.8, WFUR 1.3- CommunicatorNetwork 1.7, WMJH-AM 1.6-1.3, WJNZ-AM 1.1-1.0, WTKG-AM 1.1-1.0, WHTC- AM 0.5-0.7, WGHN 0.5-0.7, WKWM-AM 0.5-0.6, WYGR-AM 1.2-0.6, A T T L E WYVN 0.4-0.5, WMRR 0.8-0.5. Fall books found in this TATTLER are TT A T T L E RR 12+ persons, 6A-12P, M-Su, 6A-mid, Summer 2002 – Fall 2002 com- parisons, unless otherwise noted. Copyright © 2002, The Arbitron Com- TheThe intersectionintersection ofof radioradio && musicmusic sincesince 19741974 pany. These results may not be used without permission from Arbitron. TomTom KayKay -- ChrisChris MozenaMozena -- BradBrad SavageSavage The Conclave gives the 2 minute warning!! Make that, the 2 week Congrats to former Conclave Board member – and longtime Conclave warning. The Conclave wants EVERYONE to know it is STILL accepting agenda committee head – Rob Sisco as he ascends to the post of Presi- applications from high school students throughout the Upper Midwest dent, Nielsen Music and COO, Nielsen Retail Entertainment Infor- and Great Lakes region interested in studying for a career in the radio or mation (REI). -

Location Indicators by Indicator
ECCAIRS 4.2.6 Data Definition Standard Location Indicators by indicator The ECCAIRS 4 location indicators are based on ICAO's ADREP 2000 taxonomy. They have been organised at two hierarchical levels. 12 January 2006 Page 1 of 251 ECCAIRS 4 Location Indicators by Indicator Data Definition Standard OAAD OAAD : Amdar 1001 Afghanistan OAAK OAAK : Andkhoi 1002 Afghanistan OAAS OAAS : Asmar 1003 Afghanistan OABG OABG : Baghlan 1004 Afghanistan OABR OABR : Bamar 1005 Afghanistan OABN OABN : Bamyan 1006 Afghanistan OABK OABK : Bandkamalkhan 1007 Afghanistan OABD OABD : Behsood 1008 Afghanistan OABT OABT : Bost 1009 Afghanistan OACC OACC : Chakhcharan 1010 Afghanistan OACB OACB : Charburjak 1011 Afghanistan OADF OADF : Darra-I-Soof 1012 Afghanistan OADZ OADZ : Darwaz 1013 Afghanistan OADD OADD : Dawlatabad 1014 Afghanistan OAOO OAOO : Deshoo 1015 Afghanistan OADV OADV : Devar 1016 Afghanistan OARM OARM : Dilaram 1017 Afghanistan OAEM OAEM : Eshkashem 1018 Afghanistan OAFZ OAFZ : Faizabad 1019 Afghanistan OAFR OAFR : Farah 1020 Afghanistan OAGD OAGD : Gader 1021 Afghanistan OAGZ OAGZ : Gardez 1022 Afghanistan OAGS OAGS : Gasar 1023 Afghanistan OAGA OAGA : Ghaziabad 1024 Afghanistan OAGN OAGN : Ghazni 1025 Afghanistan OAGM OAGM : Ghelmeen 1026 Afghanistan OAGL OAGL : Gulistan 1027 Afghanistan OAHJ OAHJ : Hajigak 1028 Afghanistan OAHE OAHE : Hazrat eman 1029 Afghanistan OAHR OAHR : Herat 1030 Afghanistan OAEQ OAEQ : Islam qala 1031 Afghanistan OAJS OAJS : Jabul saraj 1032 Afghanistan OAJL OAJL : Jalalabad 1033 Afghanistan OAJW OAJW : Jawand 1034 -
HAS SKIP STARTED YET? for Some, It Ended in June
The Official Publication of the Worldwide TV-FM DX Association AUGUST 2013 The Magazine for TV and FM DXers IT EXISTS! Sreengrab by Chris Dunne WSBS-DT-3 KEY WEST ! Who will be the first to log it via Es or Tr? Afternoon Storm on the Plains HAS SKIP STARTED YET? For some, it ended in June. For others, it ended in early July For a few, it never even began Visit Us At www.wtfda.org THE WORLDWIDE TV-FM DX ASSOCIATION Serving the UHF-VHF Enthusiast THE VHF-UHF DIGEST IS THE OFFICIAL PUBLICATION OF THE WORLDWIDE TV-FM DX ASSOCIATION DEDICATED TO THE OBSERVATION AND STUDY OF THE PROPAGATION OF LONG DISTANCE TELEVISION AND FM BROADCASTING SIGNALS AT VHF AND UHF. WTFDA IS GOVERNED BY A BOARD OF DIRECTORS: DOUG SMITH, GREG CONIGLIO, KEITH McGINNIS AND MIKE BUGAJ. Editor and publisher: Mike Bugaj Treasurer: Keith McGinnis wtfda.org Webmaster: Tim McVey Forum Site Administrator: Chris Cervantez Editorial Staff: Jeff Kruszka, Keith McGinnis, Fred Nordquist, Nick Langan, Doug Smith, Peter Baskind, Bill Hale and John Zondlo, Website: www.wtfda.org; Forums: http://forums.wtfda.org _______________________________________________________________________________________ JULY 2013 Alan Michalek, Benjamin Greenlaw, John THAT WAS THE SEASON THAT WAS Johnson, Eugene Hinton and Stan Weisbeck. Thank you everybody for supporting your Just when you thought the skip season was WTFDA! over for the season, an opening comes along at the end of July to prove you wrong. AND MORE… As skip seasons go, this one was pretty rotten. The month of June I never mentioned this, but during the spring was almost completely void I received a phone call from Peter Oprisko, Jr. -

Investigation of Adiposity Phenotypes in AA Associated with GALNT10 & Related Pathway Genes
Investigation of Adiposity Phenotypes in AA Associated With GALNT10 & Related Pathway Genes By Mary E. Stromberg A Dissertation Submitted to the Graduate Faculty of WAKE FOREST UNIVERSITY GRADUATE SCHOOL OF ARTS AND SCIENCES in Partial Fulfillment of the Requirements for the Degree of DOCTOR OF PHILOSOPHY In Molecular Genetics and Genomics December 2018 Winston-Salem, North Carolina Approved by: Donald W. Bowden, Ph.D., Advisor Maggie C.Y. Ng, Ph.D., Advisor Timothy D. Howard, Ph.D., Chair Swapan Das, Ph.D. John P. Parks, Ph.D. Acknowledgements I would first like to thank my mentors, Dr. Bowden and Dr. Ng, for guiding my learning and growth during my years at Wake Forest University School of Medicine. Thank you Dr. Ng for spending so much time ensuring that I learn every detail of every protocol, and supporting me through personal difficulties over the years. Thank you Dr. Bowden for your guidance in making me a better scientist and person. I would like to thank my committee for their patience and the countless meetings we have had in discussing this project. I would like to say thank you to the members of our lab as well as the Parks lab for their support and friendship as well as their contributions to my project. Special thanks to Dean Godwin for his support and understanding. The umbrella program here at WFU has given me the chance to meet some of the best friends I could have wished for. I would like to also thank those who have taught me along the way and helped me to get to this point of my life, with special thanks to the late Dr. -

Tattler Master 1/21 Pm
bear by the name of Mr. Snuggly Huggily. Jay Leno mentioned it Volume XXXI • Number 3 • January 21, 2005 on his show last Friday (1/14) and the bidding topped off at THE $267.00. (plus sales tax and shipping charges) MAIN STREET The hasty January 3rd exit of Emmis WNOU/Indianapolis co-host Communicator Network Dennis “Billy Breeze” Grubbs has finally been explained in a recent article in the Indianapolis Star. Turns out Breeze aired A T T L E the phone number of BonnieJean Ventress on December 9th, TT A T T L E RR urging listeners to call the number to harass the woman, even offering a prize to the person who could give her the most trouble. Publisher • Tom Kay/Main Street Marketing & Promotion Ventresss is suing Emmis and Grubbs for pain and suffering, “All the news that fits, we gits!” claming that the calls included death threats. She also says that she had a relationship of a sexual nature with Grubbs. Neither Java Joel Murphy has been fired from Clear Channel WKSC- Emmis nor WNOU has commented on the matter. FM/Chicago. The nighttime personality made a joke about adopting “three black kids” and “taking them to the zoo to see St. Louis 2004 Fall Book. There’s Infinity N/T KMOX and AC where they came from” on January 11. Only one listener called KEZK, and then the rest. KMOX-AM 11.5-11, KEZK-FM 6.2-7.8, to complain, but that was enough to send the 30-year old WIL-FM 5.6-5.5, KMJM-FM 5.5-5, KSD-FM 4.2-4.5, KSLZ-FM personality packing the very next day. -

Molecular Evolution of the Glycosyltransferase 6 Gene Family in Primates
Hindawi Publishing Corporation Biochemistry Research International Volume 2016, Article ID 9051727, 6 pages http://dx.doi.org/10.1155/2016/9051727 Research Article Molecular Evolution of the Glycosyltransferase 6 Gene Family in Primates Eliane Evanovich,1 Patricia Jeanne de Souza Mendonça-Mattos,1 and Maria Lúcia Harada2 1 Laboratorio´ de Genetica´ Humana e Medica,´ Instituto de Cienciasˆ Biologicas,´ Universidade Federal do Para,´ Belem,´ PA, Brazil 2Laboratorio´ de Biologia Molecular “Francisco Mauro Salzano”, Instituto de Cienciasˆ Biologicas,´ Universidade Federal do Para,´ Belem,´ PA, Brazil Correspondence should be addressed to Eliane Evanovich; [email protected] Received 12 July 2016; Accepted 20 October 2016 Academic Editor: Stefano Pascarella Copyright © 2016 Eliane Evanovich et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Glycosyltransferase 6 gene family includes ABO, Ggta1, iGb3S, and GBGT1 genes and by three putative genes restricted to mammals, GT6m6, GTm6, and GT6m7, only the latter is found in primates. GT6 genes may encode functional and nonfunctional proteins. Ggta1 and GBGT1 genes, for instance, are pseudogenes in catarrhine primates, while iGb3S gene is only inactive in human, bonobo, and chimpanzee. Even inactivated, these genes tend to be conversed in primates. As some of the GT6 genes are related to the susceptibility or resistance to parasites, we investigated (i) the selective pressure on the GT6 paralogs genes in primates; (ii) the basis of the conservation of iGb3S in human, chimpanzee, and bonobo; and (iii) the functional potential of the GBGT1 and GT6m7 in catarrhines. -

2012 Community Media Directory
2012 Community Media Directory 2012 Community Media Directory A Resource Listing of Print and Broadcast Media in and around Champaign County, Illinois Updated April 2012 by Carle Foundation Hospital 1 2012 Community Media Directory Community Media Directory Table of Contents: 1. Newspapers – Champaign/Urbana - News-Gazette - News-Gazette Danville Bureau - Daily Illini - Parkland Prospectus 2. Newspapers – Regional - Arcola Record Harold - Arthur Graphic Clarion, Atwood Herald, News Record (southern Piatt Co.) and Mount Zion Region News - Bloomington Daily Pantagraph - Central Illinois Business Magazine (Monthly), Central Illinois Families Magazine (Quarterly), At Home in Central Illinois (Quarterly) - Danville Commercial News - Decatur Herald and Review - Effingham Daily News - Fisher View - Georgetown Independent News (News-Gazette Community Newspaper) - Gibson City Courier - Health Illinois Magazine - Hoopeston Chronicle - Mahomet Citizen (News-Gazette Community Newspaper) - Mattoon Journal Gazette/Times Courier - The Leader (formerly the Ogden Leader) - Paris Beacon News - Paxton Record (News-Gazette Community Newspaper) - Piatt County Journal-Republican(News-Gazette Community Newspaper) - The Rantoul Press (News-Gazette Community Newspaper) - County Star (News-Gazette Community Newspaper) - Tuscola Journal, Tri County Journal (Moultrie, Douglas, Coles counties) - UP-TV Urbana Public Television, Channel 6 3. Television - WAND-TV: NBC, Channel 17 - WBUI-TV (FOX 55/27 Illinois/Central Illinois’ CW): Channel 23 - WCCU-TV FOX (Channel 27), -
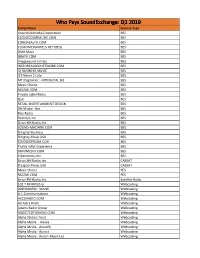
Licensee Count Q1 2019.Xlsx
Who Pays SoundExchange: Q1 2019 Entity Name License Type Aura Multimedia Corporation BES CLOUDCOVERMUSIC.COM BES COROHEALTH.COM BES CUSTOMCHANNELS.NET (BES) BES DMX Music BES GRAYV.COM BES Imagesound Limited BES INSTOREAUDIONETWORK.COM BES IO BUSINESS MUSIC BES It'S Never 2 Late BES MTI Digital Inc - MTIDIGITAL.BIZ BES Music Choice BES MUZAK.COM BES Private Label Radio BES Qsic BES RETAIL ENTERTAINMENT DESIGN BES Rfc Media - Bes BES Rise Radio BES Rockbot, Inc. BES Sirius XM Radio, Inc BES SOUND-MACHINE.COM BES Stingray Business BES Stingray Music USA BES STUDIOSTREAM.COM BES Thales Inflyt Experience BES UMIXMEDIA.COM BES Vibenomics, Inc. BES Sirius XM Radio, Inc CABSAT Stingray Music USA CABSAT Music Choice PES MUZAK.COM PES Sirius XM Radio, Inc Satellite Radio 102.7 FM KPGZ-lp Webcasting 999HANKFM - WANK Webcasting A-1 Communications Webcasting ACCURADIO.COM Webcasting Ad Astra Radio Webcasting Adams Radio Group Webcasting ADDICTEDTORADIO.COM Webcasting Aloha Station Trust Webcasting Alpha Media - Alaska Webcasting Alpha Media - Amarillo Webcasting Alpha Media - Aurora Webcasting Alpha Media - Austin-Albert Lea Webcasting Alpha Media - Bakersfield Webcasting Alpha Media - Biloxi - Gulfport, MS Webcasting Alpha Media - Brookings Webcasting Alpha Media - Cameron - Bethany Webcasting Alpha Media - Canton Webcasting Alpha Media - Columbia, SC Webcasting Alpha Media - Columbus Webcasting Alpha Media - Dayton, Oh Webcasting Alpha Media - East Texas Webcasting Alpha Media - Fairfield Webcasting Alpha Media - Far East Bay Webcasting Alpha Media