Anti-ASAP2 (Aa 5-194) Polyclonal Antibody (DPABH-00543) This Product Is for Research Use Only and Is Not Intended for Diagnostic Use
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Genetic and Genomic Analysis of Hyperlipidemia, Obesity and Diabetes Using (C57BL/6J × TALLYHO/Jngj) F2 Mice
University of Tennessee, Knoxville TRACE: Tennessee Research and Creative Exchange Nutrition Publications and Other Works Nutrition 12-19-2010 Genetic and genomic analysis of hyperlipidemia, obesity and diabetes using (C57BL/6J × TALLYHO/JngJ) F2 mice Taryn P. Stewart Marshall University Hyoung Y. Kim University of Tennessee - Knoxville, [email protected] Arnold M. Saxton University of Tennessee - Knoxville, [email protected] Jung H. Kim Marshall University Follow this and additional works at: https://trace.tennessee.edu/utk_nutrpubs Part of the Animal Sciences Commons, and the Nutrition Commons Recommended Citation BMC Genomics 2010, 11:713 doi:10.1186/1471-2164-11-713 This Article is brought to you for free and open access by the Nutrition at TRACE: Tennessee Research and Creative Exchange. It has been accepted for inclusion in Nutrition Publications and Other Works by an authorized administrator of TRACE: Tennessee Research and Creative Exchange. For more information, please contact [email protected]. Stewart et al. BMC Genomics 2010, 11:713 http://www.biomedcentral.com/1471-2164/11/713 RESEARCH ARTICLE Open Access Genetic and genomic analysis of hyperlipidemia, obesity and diabetes using (C57BL/6J × TALLYHO/JngJ) F2 mice Taryn P Stewart1, Hyoung Yon Kim2, Arnold M Saxton3, Jung Han Kim1* Abstract Background: Type 2 diabetes (T2D) is the most common form of diabetes in humans and is closely associated with dyslipidemia and obesity that magnifies the mortality and morbidity related to T2D. The genetic contribution to human T2D and related metabolic disorders is evident, and mostly follows polygenic inheritance. The TALLYHO/ JngJ (TH) mice are a polygenic model for T2D characterized by obesity, hyperinsulinemia, impaired glucose uptake and tolerance, hyperlipidemia, and hyperglycemia. -

Targeting PH Domain Proteins for Cancer Therapy
The Texas Medical Center Library DigitalCommons@TMC The University of Texas MD Anderson Cancer Center UTHealth Graduate School of The University of Texas MD Anderson Cancer Biomedical Sciences Dissertations and Theses Center UTHealth Graduate School of (Open Access) Biomedical Sciences 12-2018 Targeting PH domain proteins for cancer therapy Zhi Tan Follow this and additional works at: https://digitalcommons.library.tmc.edu/utgsbs_dissertations Part of the Bioinformatics Commons, Medicinal Chemistry and Pharmaceutics Commons, Neoplasms Commons, and the Pharmacology Commons Recommended Citation Tan, Zhi, "Targeting PH domain proteins for cancer therapy" (2018). The University of Texas MD Anderson Cancer Center UTHealth Graduate School of Biomedical Sciences Dissertations and Theses (Open Access). 910. https://digitalcommons.library.tmc.edu/utgsbs_dissertations/910 This Dissertation (PhD) is brought to you for free and open access by the The University of Texas MD Anderson Cancer Center UTHealth Graduate School of Biomedical Sciences at DigitalCommons@TMC. It has been accepted for inclusion in The University of Texas MD Anderson Cancer Center UTHealth Graduate School of Biomedical Sciences Dissertations and Theses (Open Access) by an authorized administrator of DigitalCommons@TMC. For more information, please contact [email protected]. TARGETING PH DOMAIN PROTEINS FOR CANCER THERAPY by Zhi Tan Approval page APPROVED: _____________________________________________ Advisory Professor, Shuxing Zhang, Ph.D. _____________________________________________ -

Delineation of Key Regulatory Elements Identifies Points Of
DELINEATION OF KEY REGULATORY ELEMENTS IDENTIFIES POINTS OF VULNERABILITY IN THE MITOGEN-ACTIVATED SIGNALING NETWORK SUPPLEMENTARY MATERIALS List of contents Supplementary Figures with legends 1. Figure S1: Distribution of primary siRNA screen data, and standardization of assay procedure. 2. Figure S2: Scatter plot of screen data. 3. Figure S3: Functional relevance of the identified targets and Calculation of residence time from PDT and cell cycle distribution. 4. Figure S4: FACS profiles for ABL1 and AKT1. Table for data in Figure 5B. 5. Figure S5: Venn diagram showing the results of the comparative analysis of other screen results 6. Figure S6: Dose response profiles for the AKT1 + ABL1 inhibitor combination for CH1, list of the 14 cell lines and their description, effect of ABL1+AKT1 inhibitor combination on increase in apoptotic cells and G1 arrest in 14 cell lines, effects of CHEK1 inhibitor on combination C1,C2 on 4 cell lines. Supplementary Tables 1. Table S1: siRNA screen results for targeted kinases and phosphatases. 2. Table S2: Gene expression status of the validated hits. 3. Table S3: Role played by identified RNAi hits in regulation of cell cycle, the effect on PDTs along with phase-specific RTs. 4. Table S4: List of molecules classified as cell cycle targets. 5. Table S5: High confidence network used for graph theory analysis. 6. Table S6: Occurrences of nodes in shortest path networks. 7. Table S7: Network file used as SNAVI background. 8. Table S8: Classification of nodes present in modules according to specificity. Legends for tables Supplementary Experimental Procedures References Figure S1 A 450 400 G1 S 350 G2 300 250 200 150 100 50 Distribution of molecules Distribution 0 -6-4-20246 Z-score 350 200 400 G1 S 300 G2 150 300 250 200 100 200 150 100 50 100 Distribution of molecules 50 0 0 0 -4 -2 0 2 4 -4-20246 -4-20246 Z-score B PLK1 GAPDH PLCg BTK PLCg CDC2A PLCg CHEK1 PLCg MET Distribution profiles of complete primary screen and western blots showing knockdown efficiency. -

Table 3: Average Gene Expression Profiles by Chromosome
Supplemental Data Table 1: Experimental Setup Correlation Array Reverse Fluor Array Extraction Coefficient Print Batch (Y/N) mean (range) DLD1-I.1 I A N DLD1-I.2 I B N 0.86 DLD1-I.3 I C N (0.79-0.90) DLD1-I.4 I C Y DLD1 DLD1-II.1 II D N DLD1-II.2 II E N 0.86 DLD1-II.3 II F N (0.74-0.94) DLD1-II.4 II F Y DLD1+3-II.1 II A N DLD1+3-II.2 II A N 0.85 DLD1 + 3 DLD1+3-II.3 II B N (0.64-0.95) DLD1+3-II.4 II B Y DLD1+7-I.1 I A N DLD1+7-I.2 I A N 0.79 DLD1 + 7 DLD1+7-I.3 I B N (0.68-0.90) DLD1+7-I.4 I B Y DLD1+13-I.1 I A N DLD1+13-I.2 I A N 0.88 DLD1 + 13 DLD1+13-I.3 I B N (0.84-0.91) DLD1+13-I.4 I B Y hTERT-HME-I.1 I A N hTERT-HME-I.2 I B N 0.85 hTERT-HME hTERT-HME-I.3 I C N (0.80-0.92) hTERT-HME-I.4 I C Y hTERT-HME+3-I.1 I A N hTERT-HME+3-I.2 I B N 0.84 hTERT-HME + 3 hTERT-HME+3-I.3 I C N (0.74-0.90) hTERT-HME+3-I.4 I C Y Supplemental Data Table 2: Average gene expression profiles by chromosome arm DLD1 hTERT-HME Ratio.7 Ratio.1 Ratio.3 Ratio.3 Chrom. -

Detection of Copy Number Alterations in Metastatic Melanoma by a DNA
Human Cancer Biology Detection of Copy Number Alterations in Metastatic Melanoma by aDNAFluorescenceIn situ Hybridization Probe Panel and Array Comparative Genomic Hybridization: A Southwest Oncology Group Study (S9431) Stephen R. Moore,1Diane L. Persons,2 Jeffrey A. Sosman,3 Dolores Bobadilla,1Victoria Bedell,1 David D. Smith,1Sandra R.Wolman,4 Ralph J. Tuthill,5 Jim Moon,6 Vernon K. Sondak,7 and Marilyn L. Slovak1 Abstract Purpose: Gene copy number alteration (CNA) is common in malignant melanoma and is associated with tumor development and progression. The concordance between molecular cytogenetic techniques used to determine CNA has not been evaluated on a large set of loci in malignant melanoma. Experimental Design: A panel of 16 locus-specific fluorescence in situ hybridization (FISH) probes located on eight chromosomes was used to identify CNA in touch preparations of frozen tissue samples from19 patients with metastaticmelanoma (SWOG-9431). A subset ( n =11)was analyzed using bacterial artificial chromosome (BAC) array comparative genomic hybridization (aCGH) of DNA isolated directly from touch-preparation slides. Results: By FISH, most samples showed loss near or at WISP3 /6p21, CCND3/6q22, and CDKN2A /9p21 (>75% of samples tested). More than one third of CDKN2A /9p21losses were biallelic. Gains of NEDD9/6p24, MET/7q31, and MYC/8q24 were common (57%, 47%, and 41%, respectively) and CNA events involving 9p21/7p12.3 and MET were frequently coincident, suggesting gain of the whole chromosome 7. Changes were confirmed by aCGH, which also uncovered many discreet regions of change, larger than a single BAC. Overlapping segments observed in >45% of samples included many of the loci analyzed in the FISH study, in addition to other WNT pathway members, and genes associated withTP53 pathways and DNA damage response, repair, and stability. -

DDEF2 CRISPR/Cas9 KO Plasmid (H): Sc-403676
SANTA CRUZ BIOTECHNOLOGY, INC. DDEF2 CRISPR/Cas9 KO Plasmid (h): sc-403676 BACKGROUND APPLICATIONS The Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) and DDEF2 CRISPR/Cas9 KO Plasmid (h) is recommended for the disruption of CRISPR-associated protein (Cas9) system is an adaptive immune response gene expression in human cells. defense mechanism used by archea and bacteria for the degradation of foreign genetic material (4,6). This mechanism can be repurposed for other 20 nt non-coding RNA sequence: guides Cas9 functions, including genomic engineering for mammalian systems, such as to a specific target location in the genomic DNA gene knockout (KO) (1,2,3,5). CRISPR/Cas9 KO Plasmid products enable the U6 promoter: drives gRNA scaffold: helps Cas9 identification and cleavage of specific genes by utilizing guide RNA (gRNA) expression of gRNA bind to target DNA sequences derived from the Genome-scale CRISPR Knock-Out (GeCKO) v2 library developed in the Zhang Laboratory at the Broad Institute (3,5). Termination signal Green Fluorescent Protein: to visually REFERENCES verify transfection CRISPR/Cas9 Knockout Plasmid CBh (chicken β-Actin 1. Cong, L., et al. 2013. Multiplex genome engineering using CRISPR/Cas hybrid) promoter: drives systems. Science 339: 819-823. 2A peptide: expression of Cas9 allows production of both Cas9 and GFP from the 2. Mali, P., et al. 2013. RNA-guided human genome engineering via Cas9. same CBh promoter Science 339: 823-826. Nuclear localization signal 3. Ran, F.A., et al. 2013. Genome engineering using the CRISPR-Cas9 system. Nuclear localization signal SpCas9 ribonuclease Nat. Protoc. 8: 2281-2308. -

Number 7 July 2012
VolumeVolume 16 1 -- NumberNumber 71 May -July Sept 2012ember 1997 Atlas of Genetics and Cytogenetics in Oncology and Haematology OPEN ACCESS JOURNAL INIST-CNRS Scope The Atlas of Genetics and Cytogenetics in Oncology and Haematology is a peer reviewed on-line journal in open access, devoted to genes, cytogenetics, and clinical entities in cancer, and cancer-prone diseases. It presents structured review articles ("cards") on genes, leukaemias, solid tumours, cancer-prone diseases, more traditional review articles on these and also on surrounding topics ("deep insights"), case reports in hematology, and educational items in the various related topics for students in Medicine and in Sciences. Editorial correspondance Jean-Loup Huret Genetics, Department of Medical Information, University Hospital F-86021 Poitiers, France tel +33 5 49 44 45 46 or +33 5 49 45 47 67 [email protected] or [email protected] Staff Mohammad Ahmad, Mélanie Arsaban, Jérémy Cigna, Marie-Christine Jacquemot-Perbal, Vanessa Le Berre, Anne Malo, Catherine Morel-Pair, Laurent Rassinoux, Alain Zasadzinski. Philippe Dessen is the Database Director, and Alain Bernheim the Chairman of the on-line version (Gustave Roussy Institute – Villejuif – France). The Atlas of Genetics and Cytogenetics in Oncology and Haematology (ISSN 1768-3262) is published 12 times a year by ARMGHM, a non profit organisation, and by the INstitute for Scientific and Technical Information of the French National Center for Scientific Research (INIST-CNRS) since 2008. The Atlas is hosted by INIST-CNRS (http://www.inist.fr) http://AtlasGeneticsOncology.org © ATLAS - ISSN 1768-3262 The PDF version of the Atlas of Genetics and Cytogenetics in Oncology and Haematology is a reissue of the original articles published in collaboration with the Institute for Scientific and Technical Information (INstitut de l’Information Scientifique et Technique - INIST) of the French National Center for Scientific Research (CNRS) on its electronic publishing platform I-Revues. -

Genetic-Linkage Mapping of Complex Hereditary Disorders to a Whole-Genome Molecular-Interaction Network
Downloaded from genome.cshlp.org on September 28, 2021 - Published by Cold Spring Harbor Laboratory Press Methods Genetic-linkage mapping of complex hereditary disorders to a whole-genome molecular-interaction network Ivan Iossifov,1 Tian Zheng,2 Miron Baron,3 T. Conrad Gilliam,4 and Andrey Rzhetsky4,5,6 1Department of Biomedical Informatics, Center for Computational Biology and Bioinformatics, Columbia University, New York, New York 10032, USA; 2Department of Statistics, Columbia University, New York, New York 10027, USA; 3Department of Psychiatry, Columbia University, New York, New York 10032, USA; 4Department of Human Genetics, University of Chicago, Chicago, Illinois 60637, USA; 5Department of Medicine, Institute for Genomics & Systems Biology, Computation Institute, University of Chicago, Chicago, Illinois 60637, USA Common hereditary neurodevelopmental disorders such as autism, bipolar disorder, and schizophrenia are most likely both genetically multifactorial and heterogeneous. Because of these characteristics traditional methods for genetic analysis fail when applied to such diseases. To address the problem we propose a novel probabilistic framework that combines the standard genetic linkage formalism with whole-genome molecular-interaction data to predict pathways or networks of interacting genes that contribute to common heritable disorders. We apply the model to three large genotype–phenotype data sets, identify a small number of significant candidate genes for autism (24), bipolar disorder (21), and schizophrenia (25), and -
Microarray-Based Prediction of Cytotoxicity of Tumor Cells to Arsenic Trioxide
CANCER GENOMICS & PROTEOMICS 1: 363-370 (2004) Microarray-based Prediction of Cytotoxicity of Tumor Cells to Arsenic Trioxide THOMAS EFFERTH1 and BERND KAINA2 1Center for Molecular Biology, University of Heidelberg, Im Neuenheimer Feld 282, 69120 Heidelberg; 2Institute of Toxicology, University of Mainz, Mainz, Germany Abstract. Arsenic has been used since ancient times as a the 18th and 19th century, i.e., syphilis, cancer, ulcers, etc. medicinal agent. Currently, arsenic trioxide is experiencing a (3). In the 20th century, Paul Ehrlich, the founder of thriving revival in modern oncology. The aim of this study was modern chemotherapy, found the arsenical salvarsan, which to identify the molecular predictors of sensitivity and resistance was the standard therapy against syphilis for decades (4). to arsenic trioxide. We mined the microarray database of the On the other hand, arsenic compounds can be poisonous National Cancer Institute (NCI), USA, for genes whose (5). The revival of arsenic in modern medicine was initiated expression correlated with the IC50 values for arsenic trioxide by Chinese scientists showing dramatic regression rates of of 60 cell lines of different tumor types. By COMPARE acute promyelocytic leukemia by arsenic trioxide (6). These analysis, Kendall’s Ù test, and false discovery rate (FDR) findings were subsequently corroborated in clinical studies analyses, 47 out of 9706 genes or expressed sequence tags in the U.S.A. (7). (ESTs) were identified. If the mRNA expression of the 47 Various molecular determinants of the biological effect genes or ESTs was subjected to hierarchical cluster analysis of arsenic trioxide have been elucidated. It promotes the and cluster image mapping, sensitivity or resistance of the 60 degradation of the oncogenic fusion protein of the PML and cell lines to arsenic trioxide was predictable with statistical retinoic acid receptor · (RAR·) genes which arises from significance (p=1.01 x 10-5). -

There Are 1393 Genes Significant by SAM with 90Th Percentile Confidence, the False Discovery Rate Among the 1393 Significant Genes Is 0.09968
Class 1 (NPM1 wild-type); Class 2 (NPM1 mutation). There are 1393 genes significant by SAM With 90th percentile confidence, the false discovery rate among the 1393 significant genes is 0.09968 . The delta value used to identify the significant genes is 0.78224 . The fudge factor for standard deviation is computed as 0.06115 . if ratio is >1 gene expression lower in NPM1mut 307 overlapping genes (overlap of genes lower expressed in NPM1mut and putative target genes) if ratio is <1 gene expression higher in NPM1mut rank, based Geom mean of Geom mean of Ratio of on p- ratios in class ratios in class geom Map value 1=wt 2=mut means Clone gene symbol_1 gene symbol_2 Description UG cluster Location 1 1,750 0,395 4,427 IMAGE:382787 TRH TRH TRH || Thyrotropin-releasing hormone || Hs.182231 || AA069596 || || 7200 || 98655 Hs.182231 3 MLLT3 || Myeloid/lymphoid or mixed-lineage leukemia (trithorax homolog, Drosophila); translocated to, 3 || 2 1,689 0,551 3,068 IMAGE:783998 MLLT3 MLLT3 Hs.493585 || AA443284 || || 4300 || 116155 Hs.591085 9 P2RY5 || Purinergic receptor P2Y, G-protein coupled, 5 || Hs.123464 || R91539 || purinergic receptor 3 1,866 0,497 3,754 IMAGE:196488 P2RY5 P2RY5 P2Y5=RB intorn-encoded putative G-protei || 10161 || 112029 Hs.123464 13 4 1,678 0,497 3,375 IMAGE:49923 BAALC BAALC BAALC || Brain and acute leukemia, cytoplasmic || Hs.533446 || H28986 || || 79870 || 311379 Hs.533446 8 5 1,611 0,612 2,634 IMAGE:416374 TSPAN13 TM4SF13 TM4SF13 || Tetraspanin 13 || Hs.364544 || W86201 || || 27075 || 226071 Hs.364544 7 6 1,770 0,557 3,179 -

Augmenting Bioinformatics Research with Biomedical Ontologies
Waclaw Kusnierczyk Augmenting Bioinformatics Research with Biomedical Ontologies PhD thesis 2008 Department of Computer and Information Science Faculty of Information Technology, Mathematics and Electrical Engineering Norwegian University of Science and Technology i Abstract The main objective of the reported study was to investigate how biome- dical ontologies, logically structured representations of various aspects of the biomedical reality, can help researchers in analyzing experimental data. High-throughput technologies, such as platforms for microarray gene ex- pression experiments (MAGE), yield increasingly large amounts of ‘massive’ data; successful analyses of the data require expertise in the application do- main, and to support researchers with automated methods explicit domain knowledge representations are often essential. Two attempts to construct such tools are reported here: one successful — eGOn, a web-based tool for mapping the results of microarray gene expression experiments onto the structure of the Gene Ontology; and one unsuccessful — a framework for knowledge- and case-based enhancement of biological association network- building tools. Ontologies — structured, computer-understandable accounts of expert kno- wledge — are a relatively new invention. Until only recently, biomedical ontologies were developed with little care for formal semantics, syntactic and semantic compatibility with each other, and the ontological (in a philo- sophical sense) commitments made. However, for successful integration of resources that use different ontologies to describe their content and services, integration on the ontological level is needed. While one way to achieve this is to apply some of the much-researched techniques of ontology alignment and merging, another approach is to organize the ontology creation move- ment around a common basis — a unique top-level ontology and a set of design principles. -
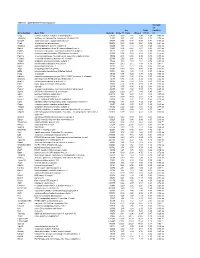
Table S-1 Mpkccd Transcriptome
Table S1. mpkCCD Cell Transcriptomes. Correlati on Ori. Ratio Coefficie Gene Symbol Gene Title GeneID Clone 11 Clone Clone 2 (11/2) nt Copg coatomer protein complex, subunit gamma 54161 1.99 1.81 2.48 0.80 -0.49 ns Atp6v0d1 ATPase, H+ transporting, lysosomal V0 subunit D1 11972 3.37 2.87 3.00 1.12 0.74 ns Golga7 golgi autoantigen, golgin subfamily a, 7 57437 3.03 3.40 3.58 0.84 -0.34 ns Psph phosphoserine phosphatase 100678 0.38 0.66 0.64 0.59 -0.61 ns Trappc4 trafficking protein particle complex 4 60409 1.17 1.12 1.08 1.08 0.64 ns Dpm2 dolichol-phosphate (beta-D) mannosyltransferase 2 13481 0.74 0.87 0.81 0.91 -0.31 ns Psmb5 proteasome (prosome, macropain) subunit, beta type 5 19173 2.92 3.00 2.92 0.99 0.32 ns Dhrs1 dehydrogenase/reductase (SDR family) member 1 52585 0.74 0.68 0.88 0.83 -0.18 ns Ppm1a protein phosphatase 1A, magnesium dependent, alpha isoform 19042 2.43 2.60 2.96 0.82 -0.53 ns Psenen presenilin enhancer 2 homolog (C. elegans) 66340 2.52 2.88 3.80 0.66 -0.77 ns Anapc1 anaphase promoting complex subunit 1 17222 1.16 1.33 1.61 0.72 -0.33 ns Mrpl43 mitochondrial ribosomal protein L43 94067 2.15 2.15 1.84 1.16 0.91 * Nmt1 N-myristoyltransferase 1 18107 3.21 2.71 3.36 0.95 0.31 ns Atg5 autophagy-related 5 (yeast) 11793 0.70 0.73 0.66 1.05 -0.23 ns Mtif2 mitochondrial translational initiation factor 2 76784 1.16 1.29 1.19 0.97 -0.36 ns Psap prosaposin 19156 8.07 7.25 8.37 0.96 0.02 ns Ube2g1 ubiquitin-conjugating enzyme E2G 1 (UBC7 homolog, C.