Synthetic Routes to New Core/Shell Nanogels
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Egyptian Literature
The Project Gutenberg EBook of Egyptian Literature This eBook is for the use of anyone anywhere at no cost and with almost no restrictions whatsoever. You may copy it, give it away or re-use it under the terms of the Project Gutenberg License included with this eBook or online at http://www.gutenberg.org/license Title: Egyptian Literature Release Date: March 8, 2009 [Ebook 28282] Language: English ***START OF THE PROJECT GUTENBERG EBOOK EGYPTIAN LITERATURE*** Egyptian Literature Comprising Egyptian Tales, Hymns, Litanies, Invocations, The Book Of The Dead, And Cuneiform Writings Edited And With A Special Introduction By Epiphanius Wilson, A.M. New York And London The Co-Operative Publication Society Copyright, 1901 The Colonial Press Contents Special Introduction. 2 The Book Of The Dead . 7 A Hymn To The Setting Sun . 7 Hymn And Litany To Osiris . 8 Litany . 9 Hymn To R ....................... 11 Hymn To The Setting Sun . 15 Hymn To The Setting Sun . 19 The Chapter Of The Chaplet Of Victory . 20 The Chapter Of The Victory Over Enemies. 22 The Chapter Of Giving A Mouth To The Overseer . 24 The Chapter Of Giving A Mouth To Osiris Ani . 24 Opening The Mouth Of Osiris . 25 The Chapter Of Bringing Charms To Osiris . 26 The Chapter Of Memory . 26 The Chapter Of Giving A Heart To Osiris . 27 The Chapter Of Preserving The Heart . 28 The Chapter Of Preserving The Heart . 29 The Chapter Of Preserving The Heart . 30 The Chapter Of Preserving The Heart . 30 The Heart Of Carnelian . 31 Preserving The Heart . 31 Preserving The Heart . -

19 Sloan-Hubert
Egypt in Antiquity: Music and Mythological Deities April Sloan–Hubert Jack Yates High School INTRODUCTION Jack Yates High School in Houston, Texas, is the alma mater of choreographer, producer and actress Debbie Allen and her Tony-award winning sister, Philicia Rashad. The Allen sisters discovered and began to develop their talents and skills at the historic Jack Yates High School. The great jazz vocalist, the late Anita Moore, developed her vocal pipes too at Jack Yates. Anita’s voice although silenced is still remembered for being hand picked by the one and only Duke Ellington to lead his orchestra. Jack Yates High School is also the alma mater of the now retired Lavonia Pope Bassett, the first African American music supervisor for the Houston Independent School District. As the present Choir Director and Fine Arts Department Chair at this talent-rich institution, I am charged with the phenomenal task to lead, mold, develop and return our department to its traditional glory. I am also cognizant of the fact that those were the days before state- mandated tests, budget cuts, site-based management and weaponless wars of destruction. In America’s not-so-long ago past, people from all walks of life were considered culturally refined and upstanding citizens by attending the opera, going to the museum, knowing what dinner fork to use at a well set table, and by the art work in their homes. At the heart of my motivation lie two exceptionally large music history classes with forty plus students each. The students enrolled in these classes are lovingly called “the music historians.” These semi-non musically inclined historians are void of vocal and instrumental skills, but they possess a great love and appreciation for music. -

Final List of Delegations
Supplément au Compte rendu provisoire (21 juin 2019) LISTE FINALE DES DÉLÉGATIONS Conférence internationale du Travail 108e session, Genève Supplement to the Provisional Record (21 June 2019) FINAL LIST OF DELEGATIONS International Labour Conference 108th Session, Geneva Suplemento de Actas Provisionales (21 de junio de 2019) LISTA FINAL DE DELEGACIONES Conferencia Internacional del Trabajo 108.ª reunión, Ginebra 2019 La liste des délégations est présentée sous une forme trilingue. Elle contient d’abord les délégations des Etats membres de l’Organisation représentées à la Conférence dans l’ordre alphabétique selon le nom en français des Etats. Figurent ensuite les représentants des observateurs, des organisations intergouvernementales et des organisations internationales non gouvernementales invitées à la Conférence. Les noms des pays ou des organisations sont donnés en français, en anglais et en espagnol. Toute autre information (titres et fonctions des participants) est indiquée dans une seule de ces langues: celle choisie par le pays ou l’organisation pour ses communications officielles avec l’OIT. Les noms, titres et qualités figurant dans la liste finale des délégations correspondent aux indications fournies dans les pouvoirs officiels reçus au jeudi 20 juin 2019 à 17H00. The list of delegations is presented in trilingual form. It contains the delegations of ILO member States represented at the Conference in the French alphabetical order, followed by the representatives of the observers, intergovernmental organizations and international non- governmental organizations invited to the Conference. The names of the countries and organizations are given in French, English and Spanish. Any other information (titles and functions of participants) is given in only one of these languages: the one chosen by the country or organization for their official communications with the ILO. -

Prayers of Renunciation EGYPTIAN GODS
Prayers of Renunciation: EGYPTIAN GODS Ephesians 6:10-12 “10 Finally, my brethren, be strong in the Lord, and in the power of his might. 11 Put on the whole armour of GOD, that ye may be able to stand against the wiles of the devil. 12 For we wrestle not against lesh and blood, but against principalities, against powers, against the rulers of the darkness of this world, against spiritual wickedness in high places.” Amanda Buys’ Spiritual Covering This is a product of Kanaan Ministries, a non-profit ministry under the covering of: • Roly, Amanda’s husband for more than thirty-five years. • River of Life Family Church Pastor Edward Gibbens Vanderbijlpark South Africa Tel: +27 (0) 16 982 3022 Fax: +27 (0) 16 982 2566 Email: [email protected] There is no copyright on this material. However, no part may be reproduced and/or presented for personal gain. All rights to this material are reserved to further the Kingdom of our Lord Jesus Christ ONLY. For further information or to place an order, please contact us at: P.O. Box 15253 27 John Vorster Avenue Panorama Plattekloof Ext. 1 7506 Panorama 7500 Cape Town Cape Town South Africa South Africa Tel: +27 (0) 21 930 7577 Fax: 086 681 9458 E-mail: [email protected] Website: www.kanaanministries.org Office hours: Monday to Friday, 9 AM to 3 PM Kanaan International Website Website: www.eu.kanaanministries.org 2 contents Preface ... 5 Declaration of confidence in GOD’s Protection ... 8 Sealing-off prayer before deliverance ... 9 1) Egyptian deities .. -
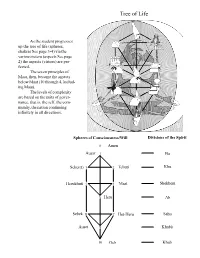
Metu Neter, Vol 1 and 2, the Afrocentric Guide to a Spiritual Union, and the Tree of Life Meditation System, by Ra Un Nefer Amen the Divisions of the Spirit
Tree of Life As the student progresses up the tree of life (spheres, chakras See page 3-4) via the various neteru (aspects See page 2) the aspects (virtues) are per- fected. The seven principles of Maat, then, become the aspects below Maat (10 through 4, includ- ing Maat). The levels of complexity are based on the units of gover- nance, that is, the self, the com- munity, the nation continuing infinitely in all directions. Spheres of Consciousness/Will Divisions of the Spirit 0 Amen Ausar 1 Ba Seker(t) 3 2 Tehuti Khu Herukhuti 5 4 Maat Shekhem 6 Heru Ab Sebek 87Het-Heru Sahu Auset 9 Khabit 10 Geb Khab 0 Amen Be able to remain peaceful in the face of the greatest difficulties. It is concerned with the issue of happiness. 1 Ausar Be able to unify all aspects of our lives; there must be unity between all of our beliefs, emotions, and actions; we must be in unity with nature and all other people. This is the chief determinant of prosperity and peace in the world, and it is accomplished through the fulfillment of the spiritual requirements for bringing God into the forefront of our consciousness to rule as our true Self. 2 Tehuti Be able to avoid and solve all conflicts, especially where there are no precedents to guide us. This is accomplished through the fulfillment of the spiritual requirements that enables the manifestation of Gods wisdom through our minds. 3 Seker Be able to overcome all difficulties and accomplish all necessary objectives, especially where there are no external means of doing so. -

Ten Egyptian Plagues for Ten Egyptian Gods and Goddesses the God of Israel Is Greater Than All Other Egyptian Gods and Goddesses
- Ten Egyptian Plagues For Ten Egyptian Gods and Goddesses The God of Israel is greater than all other Egyptian Gods and Goddesses. Moses was a great prophet, called by God with a very important job to do. As an instrument in the Lord's hand he performed many signs, or "wonders", attempting to convince Pharaoh to allow the Israelites freedom from their bondage of slavery to the Egyptians. These "wonders" are more commonly referred to as "plagues" sent from the God of Israel, as a proof that the "one true God" was far greater than all of the multiple Gods of the Egyptians. These Egyptian Plagues were harsh and varied to correspond to the ancient egyptian gods and goddesses that were prevelant during Moses time in Egypt. The number ten is a significant number in biblical numerology. It represents a fullness of quantity. Ten Egyptian Plagues Means Completely Plagued. Just as the "Ten Commandments" become symbolic of the fullness of the moral law of God, the ten ancient plagues of Egypt represent the fullness of God's expression of justice and judgments, upon those who refuse to repent. Ten times God, through Moses, allows Pharaoh to change his mind, repent, and turn to the one true God, each time increasing the severity of the consequence of the plagues suffered for disobedience to His request. Ten times Pharaoh, because of pride, refuses to be taught by the Lord, and receives "judgments" through the plagues, pronounced upon his head from Moses, the deliverer. The Ten Egyptian Plagues testify of Jesus the Anointed One and His power to save. -
A Critical Analysis of the Mytho-Reality Complexity of the Azanian Nation
Azanism: A Critical Analysis of the Mytho-Reality Complexity of the Azanian Nation Dissertation zur Erlangung des Doktorgrades an der Fakultaet Wirtschafts- und Sozialwissenschaften, Fachbereich Sozialwissenschaften der Universitaet Hamburg Vorgelegt von: Raul Guevara Diaz October 2009 Angaben der Gutachter Erste Gutachter: Zweitgutachterin Professor Dr. Cord Jakobeit Prof. Dr. Marienne Pieper Institut fuer Politikwissenschaft, Institut fuer Soziologie, Allende-Platz 1, Allende-Platz 1, 20146 Hamburg, 20146 Hamburg, Deutschland Deutschland Datum der letztzen muendlichen Pruefung 19 Mai, 2011 1 I. INTRODUCTION Substantial amount of academic literature in the field of social sciences (specialized in ethnic and nationalist politics) has dealt considerably with both the colonial and post-colonial aspects of the social and political history of Africa, and undeniably the conventional wisdom about Africa‘s political landscape should be best characterized as enduring instability. Two main factors, namely the role of colonialism and the [supposed] heterogeneity of the society, are considered crucial to explaining such a disturbing socio-political scenario. As would be expected most concern scholars and authors in this field have dealt with the general political situation in Africa within the modern paradigm of territorial nation-states. In other words, most theories of ethnic and nationalist politics have dealt with Africa‘s political instability within the formal context of the national state system (or statism). Even those who have attempted to explore the possibility of an integrated or homogeneous social growth or identity formation prior to indigenous Africans encounter with colonialism have often done so within that modern paradigm of statism. Hence, unsurprisingly, conventional wisdom espoused specifically by agents of colonialism/pseudo-nationalism tends to consider Africa‘s different dialects or linguistic groups as constituting ethnic and/or national categories in their own right. -
The 11 Laws of Amen *Paut Neteru, Tree of Life*
THE 11 LAWS OF AMEN *PAUT NETERU, TREE OF LIFE* 1 I. Law of Amen You were made in the likeness of a peace that nothing can disturb. Reclaim your peace that you may attain to your reason for coming into existence – the enjoyment of life. Reasoning: If in truth it is our nature to be at peace (free of automatic emotional responses) in situations of challenge, then the only thing we need to do is to ignore the emotional reflexes that come up in such situations. What is the point of suffering and destroying our health and performance abilities if we can be at peace—especially when the peace in situations of challenge leads to enhanced intuition and spiritual power. Amen Truisms: I live expecting neither gain nor loss, pain nor pleasure from the things I need in life, because my nature is essentially unconditioned. That which is my Self has no likes, dislikes, preferences or predetermined emotional or thought responses to situations. I am essentially unconditioned. I cultivate my happiness through spiritual development. I understand that happiness is not a continuous freedom from pain resulting from difficulties. No one knows my name, neither men nor gods. No one has seen my face, neither my father nor my mother. I was before the first time and shall be beyond the last. * 2 II. Law of Ausar Your nature is unconquerable peace, therefore nothing or no one in the world can be against you. All experiences come to you to promote your reclamation of peace, that you may in turn acquire wisdom and spiritual power. -

Legends of the Gods
LEGENDS OF THE GODS The Egyptian Texts, edited with Translations by E. A. Wallis Budge London, 1912 [Editorial note: Throughout the text "####" represents images which cannot be transcribed.] PREFACE The welcome which has been accorded to the volumes of this Series, and the fact that some of them have passed into second and third editions, suggest that these little books have been found useful by beginners in Egyptology and others. Hitherto the object of them has been to supply information about the Religion, Magic, Language, and History of the ancient Egyptians, and to provide editions of the original texts from which such information was derived. There are, however, many branches of Egyptology which need treatment in a similar manner in this Series, and it has been suggested in many quarters that the time has now arrived when the publication of a series of groups of texts illustrating Egyptian Literature in general might well be begun. Seeing that nothing is known about the authors of Egyptian works, not even their names, it is impossible to write a History of Egyptian Literature in the ordinary sense of the word. The only thing to be done is to print the actual works in the best and most complete form possible, with translations, and then to put them in the hands of the reader and leave them to his judgment. With this object in view, it has been decided to publish in the Series several volumes which shall be devoted to the reproduction in hieroglyphic type of the best and most typical examples of the various kinds of Egyptian Literature, with English translations, on a much larger scale than was possible in my "First Steps in Egyptian" or in my "Egyptian Reading Book." These volumes are intended to serve a double purpose, i.e., to supply the beginner in Egyptian with new material and a series of reading books, and to provide the general reader with translations of Egyptian works in a handy form. -

Egyptian Ideas of the Future Life
Egyptian Ideas of the Future Life by E. A. Wallis Budge. M.A., Litt.D, D.Lit. Keeper of the Egyptian and Assyrian Antiquities at the British Museum (1908). London: Kegan Paul, Trench, Tr’ubner & Co., Ltd, 1899 Preface The following pages are intended to place before the reader in a handy form an account of the principal ideas and beliefs held by the ancient Egyptians concerning the resurrection and the future life, which is derived wholly from native religious works. The literature of Egypt which deals with these subjects is large and, as was to be expected, the product of different periods which, taken together, cover several thousands of years; and it is exceedingly difficult at times to reconcile the statements and beliefs of a writer of one period with those of a writer of another. Up to the present no systematic account of the doctrine of the resurrection and of the future life has been discovered, and there is no reason for hoping that such a thing will ever be found, for the Egyptians do not appear to have thought that it was necessary to write a work of the kind. The inherent difficulty of the subject, and the natural impossibility that different men living in different places and at different times should think alike on matters which must, after all, belong always to the region of faith, render it more than probable that no college of priests, however powerful, was able to formulate a system of beliefs which would be received throughout Egypt by the clergy and the laity alike, and would be copied by the scribes as a final and authoritative work on Egyptian eschatology. -

India As Ideal and Image in Konstantin Bal'mont's
THE LAND OF THOUGHT: INDIA AS IDEAL AND IMAGE IN KONSTANTIN BAL’MONT’S OEUVRE DISSERTATION Presented in Partial Fulfillment of the Requirements of the Degree Doctor of Philosophy in the Graduate School of The Ohio State University By Susmita Sundaram * * * * * The Ohio State University 2004 Doctoral Examination Committee: Professor Irene Masing-Delic, Advisor Approved by Professor George Kalbouss Professor Yana Hashamova Professor Katya Hokanson Adviser Department of Slavic and East European Languages and Literatures A NOTE ON TRANSLATION AND TRANSLITERATION All original quotes from Russian have been transliterated with translations immediately following. The transliteration is done according to the Library of Congress system. All translations are done by the author unless otherwise indicated. ABSTRACT Russian writers have grappled with the notion of Russian identity between East and West and have mostly looked to Europe for answers since Peter the Great’s reforms. The discussion of Russia’s identity and historically ordained mission in the world came into sharp focus with the Slavophile-Westernizer debate in the first half of the nineteenth century, the resonance from which have informed Russian cultural philosophy since. Dostoevsky’s “Pushkin speech” in 1881 introduced the notion of Pushkin as a “Universal Poet” who could transcend national boundaries as a result of his universal cultural receptivity and yet could at the same time remain quintessentially Russian. The Russian Silver Age (1890’s-1910) witnessed a revival of the debate over Russia’s mission in a crisis-ridden fin-de-siècle Europe and Russia. Russian Symbolist writers looked to other cultures – in particular classical antiquity, and renaissance Italy – for cultural models that would provide an insight into solving the crisis of positivism and naturalism. -

Contextualizing the Osiris Shaft at Rosetau (Giza) in Archaeological History
University of Montana ScholarWorks at University of Montana Graduate Student Theses, Dissertations, & Professional Papers Graduate School 2021 The Lost Histories of the Shetayet of Sokar: Contextualizing the Osiris Shaft at Rosetau (Giza) in Archaeological History Nicholas Edward Whiting University of Montana, Missoula Follow this and additional works at: https://scholarworks.umt.edu/etd Part of the Archaeological Anthropology Commons, Folklore Commons, and the Social and Cultural Anthropology Commons Let us know how access to this document benefits ou.y Recommended Citation Whiting, Nicholas Edward, "The Lost Histories of the Shetayet of Sokar: Contextualizing the Osiris Shaft at Rosetau (Giza) in Archaeological History" (2021). Graduate Student Theses, Dissertations, & Professional Papers. 11693. https://scholarworks.umt.edu/etd/11693 This Thesis is brought to you for free and open access by the Graduate School at ScholarWorks at University of Montana. It has been accepted for inclusion in Graduate Student Theses, Dissertations, & Professional Papers by an authorized administrator of ScholarWorks at University of Montana. For more information, please contact [email protected]. The Lost Histories of the Shetayet of Sokar: Contextualizing the Osiris Shaft at Rosetau (Giza) in Archaeological History By Nicholas Edward Whiting Bachelor’s Degree in Anthropology, University of Montana, Missoula, Montana, 2015 Bachelor’s Degree in Legal Studies, Kaplan University, Chicago, Illinois, 2009 Thesis presented in partial fulfillment of the requirements for the degree of Master of Arts in Anthropology/Archaeology Option The University of Montana Missoula, MT Official Graduation Date January 2021 Approved by: Scott Whittenburg, Dean of The Graduate School Graduate School Dr. Kelly Dixon, Chair Professor of Anthropology Dr.