Sex Differentiation and a Chromosomal Inversion Lead to Cryptic Diversity In
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Freshwater Herring of Lake Tanganyika Are the Product of a Marine Invasion Into West Africa
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Open Marine Archive Marine Incursion: The Freshwater Herring of Lake Tanganyika Are the Product of a Marine Invasion into West Africa Anthony B. Wilson1,2¤*, Guy G. Teugels3, Axel Meyer1 1 Department of Biology, University of Konstanz, Konstanz, Germany, 2 Zoological Museum, University of Zurich, Zurich, Switzerland, 3 Ichthyology Laboratory, Royal Museum for Central Africa, Tervuren, Belgium Abstract The spectacular marine-like diversity of the endemic fauna of Lake Tanganyika, the oldest of the African Great Lakes, led early researchers to suggest that the lake must have once been connected to the ocean. Recent geophysical reconstructions clearly indicate that Lake Tanganyika formed by rifting in the African subcontinent and was never directly linked to the sea. Although the Lake has a high proportion of specialized endemics, the absence of close relatives outside Tanganyika has complicated phylogeographic reconstructions of the timing of lake colonization and intralacustrine diversification. The freshwater herring of Lake Tanganyika are members of a large group of pellonuline herring found in western and southern Africa, offering one of the best opportunities to trace the evolutionary history of members of Tanganyika’s biota. Molecular phylogenetic reconstructions indicate that herring colonized West Africa 25–50MYA, at the end of a major marine incursion in the region. Pellonuline herring subsequently experienced an evolutionary radiation in West Africa, spreading across the continent and reaching East Africa’s Lake Tanganyika during its early formation. While Lake Tanganyika has never been directly connected with the sea, the endemic freshwater herring of the lake are the descendents of an ancient marine incursion, a scenario which may also explain the origin of other Tanganyikan endemics. -

Fish, Various Invertebrates
Zambezi Basin Wetlands Volume II : Chapters 7 - 11 - Contents i Back to links page CONTENTS VOLUME II Technical Reviews Page CHAPTER 7 : FRESHWATER FISHES .............................. 393 7.1 Introduction .................................................................... 393 7.2 The origin and zoogeography of Zambezian fishes ....... 393 7.3 Ichthyological regions of the Zambezi .......................... 404 7.4 Threats to biodiversity ................................................... 416 7.5 Wetlands of special interest .......................................... 432 7.6 Conservation and future directions ............................... 440 7.7 References ..................................................................... 443 TABLE 7.2: The fishes of the Zambezi River system .............. 449 APPENDIX 7.1 : Zambezi Delta Survey .................................. 461 CHAPTER 8 : FRESHWATER MOLLUSCS ................... 487 8.1 Introduction ................................................................. 487 8.2 Literature review ......................................................... 488 8.3 The Zambezi River basin ............................................ 489 8.4 The Molluscan fauna .................................................. 491 8.5 Biogeography ............................................................... 508 8.6 Biomphalaria, Bulinis and Schistosomiasis ................ 515 8.7 Conservation ................................................................ 516 8.8 Further investigations ................................................. -

FISH4ACP Unlocking the Potential of Sustainable Fisheries and Aquaculture in Africa, the Caribbean and the Pacific
FISH4ACP Unlocking the potential of sustainable fisheries and aquaculture in Africa, the Caribbean and the Pacific United Republic of Tanzania FISH4ACP aims to strengthen and safeguard the sardine, sprat and perch value chains in Lake Tanganyika by investing in inclusive growth WHAT WE focus to bolster food security for future generations, reduce poverty and on contribute to the conservation of natural resources. → Comprehensive value chain analysis to make Lake VALUE CHAIN AT A GLANCE Tanganyika sardine, sprat and perch sector more Lake Tanganyika sardine (Limnothrissa miodon), sustainable. sprat (Stolothrissa tanganicae) and perch (Lates stappersii) → Enhancement of fish handling and processing methods to limit post-harvest losses and boost fish product quality. → Reduce harmful health impacts of fish smoking and accelerate energy use efficiency by expanding best practice fish-smoking techniques and using kilns. PRODUCTION METHOD VOLUMES * VALUE * → Capacity building on sustainable fishing practices Night fishing Artisanal. Sprat: 30 995 and use of gear. with lights, lamps and USD 117 lift-nets (sprat and sardine). Sardine: 6 315 Vertical handlines, Perch: 22 264 million → Strengthen value chain jigged lines, gill-nets and tonnes governance and empower lift-nets (perch) institutions for sustainable Source: Food and Agriculture Organization of the United Nations, fisheries management. Original Scientific Illustrations Archive. Reproduced with permission Facts figures The& United Republic of Tanzania is Lake Tanganyika’s principal producer of sardine, sprat and perch, accounting for up to 40% of the annual catch. Exports of Lake Tanganyika sprat, sardine and perch from the United Republic of Tanzania were ©FAO Hashim Muumin ©FAO worth USD 1.1 million in 2018. -

IN Lake KARIBA ~As Development Ad Ndon)
• View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Aquatic Commons THE A leA JOU L OF (Afr. J Trop. Hydrobiol. Fish) Yo. 1 1994 ASPECTS OF THE BIOLOGY OF THE lAKE TANGANYIKA SARDINE, LIMNOTHRISSA n illustrated key to the flinji. (Mimeographed MIODON (BOULENGER), IN lAKE KARIBA ~as Development Ad ndon). R. HUDDART ision des Synodontis Kariba Research Unit, Private Bag 24 CX, CHOMA, Zambia. e Royal de I'Afrique 'en, Belgium. 500p. Present address:- ~). The ecology of the 4, Ingleside Grove, LONDON, S.B.3., U.K. is (Pisces; Siluoidea) Nigeria. (Unpublished ABSTRACT o the University of Juveniles of U11lnothrissa 11liodon (Boulenger) were introduced into the man-made Lake Kariba in ngland). 1967-1968. Thirty months of night-fishing for this species from Sinazongwe, near the centre of the Kariba North bank. from 1971 to 1974 are described. Biological studies were carried out on samples of the catch during most of these months. Limnological studies were carried out over a period of four months in 1973. Li11lnothrissa is breeding successfully and its number have greatly increased. [t has reached an equilibrium level of population size at a [ower density than that of Lake Tanganyika sardines, but nevertheless is an important factor in the ecology of Lake Kariba. The growth rate, size at maturity and maximum size are all less than those of Lake Tanganyika Li11lnothrissa. A marked disruption in the orderly progression of length frequency modes occurs in September, for which the present body of evidence cannot supply an explanation. INTRODUCTION light-attracted sardines in Lake Kariba were The absence of a specialised, plankti tested in 1970 and early 1971. -

Thezambiazimbabwesadc Fisheriesprojectonlakekariba: Reportfroma Studytnp
279 TheZambiaZimbabweSADC FisheriesProjectonLakeKariba: Reportfroma studytnp •TrygveHesthagen OddTerjeSandlund Tor.FredrikNæsje TheZambia-ZimbabweSADC FisheriesProjectonLakeKariba: Reportfrom a studytrip Trygve Hesthagen Odd Terje Sandlund Tor FredrikNæsje NORSKINSTITI= FORNATURFORSKNNG O Norwegian institute for nature research (NINA) 2010 http://www.nina.no Please contact NINA, NO-7485 TRONDHEIM, NORWAY for reproduction of tables, figures and other illustrations in this report. nina oppdragsmelding279 Hesthagen,T., Sandlund, O.T. & Næsje, T.F. 1994. NINAs publikasjoner The Zambia-Zimbabwe SADC fisheries project on Lake Kariba: Report from a study trip. NINA NINA utgirfem ulikefaste publikasjoner: Oppdragsmelding279:1 17. NINA Forskningsrapport Her publiseresresultater av NINAs eget forskning- sarbeid, i den hensiktå spre forskningsresultaterfra institusjonen til et større publikum. Forsknings- rapporter utgis som et alternativ til internasjonal Trondheimapril 1994 publisering, der tidsaspekt, materialets art, målgruppem.m. gjør dette nødvendig. ISSN 0802-4103 ISBN 82-426-0471-1 NINA Utredning Serien omfatter problemoversikter,kartlegging av kunnskapsnivået innen et emne, litteraturstudier, sammenstillingav andres materiale og annet som ikke primært er et resultat av NINAs egen Rettighetshaver0: forskningsaktivitet. NINA Norskinstituttfornaturforskning NINA Oppdragsmelding Publikasjonenkansiteresfritt med kildeangivelse Dette er det minimum av rapporteringsomNINA gir til oppdragsgiver etter fullført forsknings- eller utredningsprosjekt.Opplageter -

Development of Some Lake Ecosystems in Tropical Africa, with Special Reference to the Invertebrates
Biol. Rev. ( 1974), 49. PP- 365-397 365 BRC PAH 49-10 DEVELOPMENT OF SOME LAKE ECOSYSTEMS IN TROPICAL AFRICA, WITH SPECIAL REFERENCE TO THE INVERTEBRATES By A. J. M cL A C H L A N Zoology Department, University of Newcastle upon Tyne NE 1 7RU, Britain {Received 22 January 1974) CONTENTS I n t r o d u c t i o n .......................................................................................................... 365 Man made lakes ........... 369 Lakes Kariba and Volta . 371 The pre-impoundment catchment . .371 The filling p h a s e .................................................................................................372 (а) Physical and chemical characteristics ...... 372 (i) Turbidity .......... 372 (ii) Nutrient salts .......... 372 (iii) O x y g e n .................................................................................................373 (iv) Organic matter ......... 373 (б) Animals and p la n t s ............................................................................. 374 (i) Explosive population growth ....... 374 (ii) Distribution p a t t e r n s ....................................................................375 (c) Conclusions ........... 378 The post-filling phase .......... 378 (a) The role of submerged woodland ....... 380 (b) Development of the mud habitat ....... 382 (c) Rooted aquatic plants as a habitat for sedentary animals . 384 (d) Fluctuations in lake level ........ 385 (e) Conclusions ........... 388 Equilibrium p h a s e .................................................................................................388 -

Food Resources of Lake Tanganyika Sardines Metabarcoding of the Stomach Content of Limnothrissa Miodon and Stolothrissa Tanganicae
FACULTY OF SCIENCE Food resources of Lake Tanganyika sardines Metabarcoding of the stomach content of Limnothrissa miodon and Stolothrissa tanganicae Charlotte HUYGHE Supervisor: Prof. F. Volckaert Thesis presented in Laboratory of Biodiversity and Evolutionary Genomics fulfillment of the requirements Mentor: E. De Keyzer for the degree of Master of Science Laboratory of Biodiversity and Evolutionary in Biology Genomics Academic year 2018-2019 © Copyright by KU Leuven Without written permission of the promotors and the authors it is forbidden to reproduce or adapt in any form or by any means any part of this publication. Requests for obtaining the right to reproduce or utilize parts of this publication should be addressed to KU Leuven, Faculteit Wetenschappen, Geel Huis, Kasteelpark Arenberg 11 bus 2100, 3001 Leuven (Heverlee), Telephone +32 16 32 14 01. A written permission of the promotor is also required to use the methods, products, schematics and programs described in this work for industrial or commercial use, and for submitting this publication in scientific contests. i ii Acknowledgments First of all, I would like to thank my promotor Filip for giving me this opportunity and guiding me through the thesis. A very special thanks to my supervisor Els for helping and guiding me during every aspect of my thesis, from the sampling nights in the middle of Lake Tanganyika to the last review of my master thesis. Also a special thanks to Franz who helped me during the lab work and statistics but also guided me throughout the thesis. I am very grateful for all your help and advice during the past year. -
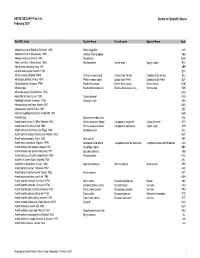
ASFIS ISSCAAP Fish List February 2007 Sorted on Scientific Name
ASFIS ISSCAAP Fish List Sorted on Scientific Name February 2007 Scientific name English Name French name Spanish Name Code Abalistes stellaris (Bloch & Schneider 1801) Starry triggerfish AJS Abbottina rivularis (Basilewsky 1855) Chinese false gudgeon ABB Ablabys binotatus (Peters 1855) Redskinfish ABW Ablennes hians (Valenciennes 1846) Flat needlefish Orphie plate Agujón sable BAF Aborichthys elongatus Hora 1921 ABE Abralia andamanika Goodrich 1898 BLK Abralia veranyi (Rüppell 1844) Verany's enope squid Encornet de Verany Enoploluria de Verany BLJ Abraliopsis pfefferi (Verany 1837) Pfeffer's enope squid Encornet de Pfeffer Enoploluria de Pfeffer BJF Abramis brama (Linnaeus 1758) Freshwater bream Brème d'eau douce Brema común FBM Abramis spp Freshwater breams nei Brèmes d'eau douce nca Bremas nep FBR Abramites eques (Steindachner 1878) ABQ Abudefduf luridus (Cuvier 1830) Canary damsel AUU Abudefduf saxatilis (Linnaeus 1758) Sergeant-major ABU Abyssobrotula galatheae Nielsen 1977 OAG Abyssocottus elochini Taliev 1955 AEZ Abythites lepidogenys (Smith & Radcliffe 1913) AHD Acanella spp Branched bamboo coral KQL Acanthacaris caeca (A. Milne Edwards 1881) Atlantic deep-sea lobster Langoustine arganelle Cigala de fondo NTK Acanthacaris tenuimana Bate 1888 Prickly deep-sea lobster Langoustine spinuleuse Cigala raspa NHI Acanthalburnus microlepis (De Filippi 1861) Blackbrow bleak AHL Acanthaphritis barbata (Okamura & Kishida 1963) NHT Acantharchus pomotis (Baird 1855) Mud sunfish AKP Acanthaxius caespitosa (Squires 1979) Deepwater mud lobster Langouste -

Diversity, Origin and Intra- Specific Variability
Contributions to Zoology, 87 (2) 105-132 (2018) Monogenean parasites of sardines in Lake Tanganyika: diversity, origin and intra- specific variability Nikol Kmentová1, 15, Maarten Van Steenberge2,3,4,5, Joost A.M. Raeymaekers5,6,7, Stephan Koblmüller4, Pascal I. Hablützel5,8, Fidel Muterezi Bukinga9, Théophile Mulimbwa N’sibula9, Pascal Masilya Mulungula9, Benoît Nzigidahera†10, Gaspard Ntakimazi11, Milan Gelnar1, Maarten P.M. Vanhove1,5,12,13,14 1 Department of Botany and Zoology, Faculty of Science, Masaryk University, Kotlářská 2, 611 37 Brno, Czech Republic 2 Biology Department, Royal Museum for Central Africa, Leuvensesteenweg 13, 3080, Tervuren, Belgium 3 Operational Directorate Taxonomy and Phylogeny, Royal Belgian Institute of Natural Sciences, Vautierstraat 29, B-1000 Brussels, Belgium 4 Institute of Biology, University of Graz, Universitätsplatz 2, A-8010 Graz, Austria 5 Laboratory of Biodiversity and Evolutionary Genomics, Department of Biology, University of Leuven, Ch. Deberiotstraat 32, B-3000 Leuven, Belgium 6 Centre for Biodiversity Dynamics, Department of Biology, Norwegian University of Science and Technology, N-7491 Trondheim, Norway 7 Faculty of Biosciences and Aquaculture, Nord University, N-8049 Bodø, Norway 8 Flanders Marine Institute, Wandelaarkaai 7, 8400 Oostende, Belgium 9 Centre de Recherche en Hydrobiologie, Département de Biologie, B.P. 73 Uvira, Democratic Republic of Congo 10 Office Burundais pour la Protection de l‘Environnement, Centre de Recherche en Biodiversité, Avenue de l‘Imprimerie Jabe 12, B.P. -

Masaryk University Faculty of Science
MASARYK UNIVERSITY FACULTY OF SCIENCE The parasite fauna of economically important pelagic fishes in Lake Tanganyika Ph.D. Thesis NIKOL KMENTOVÁ Supervisor: Maarten Vanhove, Ph.D. Department of Botany and Zoology Brno 2019 Bibliographic Entry Author Mgr. Nikol Kmentová Faculty of Science, Masaryk University Department of Botany and Zoology Title of Thesis: The parasite fauna of economically important pelagic fishes in Lake Tanganyika Degree programme: Ecological and Evolutionary Biology Specialization: Parasitology Supervisor: Maarten Vanhove, Ph.D. Academic Year: 2019/2020 Number of Pages: 350 + 72 Keywords: Kapentagyrus, Dolicirrolectanum, Cryptogonimidae, Clupeidae, Latidae, Bathybatini Bibliografický záznam Autor: Mgr. Nikol Kmentová Přírodovědecká fakulta, Masarykova univerzita Ústav botaniky a zoologie Název práce: Parazité ekonomicky významných ryb pelagické zóny jezera Tanganika Studijní program: Ekologická a evoluční biologie Specializace: Parazitologie Vedoucí práce: Maarten Vanhove, Ph.D. Akademický rok: 2019/2020 Počet stran: 350 + 72 Klíčová slova: Kapentagyrus, Dolicirrolectanum, Cryptogonimidae, Clupeidae, Latidae, Bathybatini ABSTRACT Biodiversity is a well-known term characterising the variety and variability of life on Earth. It consists of many different levels with species richness as the most frequently used measure. Despite its generally lower species richness compared to littoral zones, the global importance of the pelagic realm in marine and freshwater ecosystems lies in the high level of productivity supporting fisheries worldwide. In terms of endemicity, Lake Tanganyika is one of the most exceptional freshwater study areas in the world. While dozens of studies focus on this lake’s cichlids as model organisms, our knowledge about the economically important fish species is still poor. Despite their important role in speciation processes, parasite taxa have been vastly ignored in the African Great Lakes including Lake Tanganyika for many years. -

Vital but Vulnerable: Climate Change Vulnerability and Human Use of Wildlife in Africa’S Albertine Rift
Vital but vulnerable: Climate change vulnerability and human use of wildlife in Africa’s Albertine Rift J.A. Carr, W.E. Outhwaite, G.L. Goodman, T.E.E. Oldfield and W.B. Foden Occasional Paper for the IUCN Species Survival Commission No. 48 The designation of geographical entities in this book, and the presentation of the material, do not imply the expression of any opinion whatsoever on the part of IUCN or the compilers concerning the legal status of any country, territory, or area, or of its authorities, or concerning the delimitation of its frontiers or boundaries. The views expressed in this publication do not necessarily reflect those of IUCN or other participating organizations. Published by: IUCN, Gland, Switzerland Copyright: © 2013 International Union for Conservation of Nature and Natural Resources Reproduction of this publication for educational or other non-commercial purposes is authorized without prior written permission from the copyright holder provided the source is fully acknowledged. Reproduction of this publication for resale or other commercial purposes is prohibited without prior written permission of the copyright holder. Citation: Carr, J.A., Outhwaite, W.E., Goodman, G.L., Oldfield, T.E.E. and Foden, W.B. 2013. Vital but vulnerable: Climate change vulnerability and human use of wildlife in Africa’s Albertine Rift. Occasional Paper of the IUCN Species Survival Commission No. 48. IUCN, Gland, Switzerland and Cambridge, UK. xii + 224pp. ISBN: 978-2-8317-1591-9 Front cover: A Burundian fisherman makes a good catch. © R. Allgayer and A. Sapoli. Back cover: © T. Knowles Available from: IUCN (International Union for Conservation of Nature) Publications Services Rue Mauverney 28 1196 Gland Switzerland Tel +41 22 999 0000 Fax +41 22 999 0020 [email protected] www.iucn.org/publications Also available at http://www.iucn.org/dbtw-wpd/edocs/SSC-OP-048.pdf About IUCN IUCN, International Union for Conservation of Nature, helps the world find pragmatic solutions to our most pressing environment and development challenges. -

Masaryk University Faculty of Science
MASARYK UNIVERSITY FACULTY OF SCIENCE The parasite fauna of economically important pelagic fishes in Lake Tanganyika Ph.D. Thesis NIKOL KMENTOVÁ Supervisor: Maarten Vanhove, Ph.D. Department of Botany and Zoology Brno 2019 Bibliographic Entry Author Mgr. Nikol Kmentová Faculty of Science, Masaryk University Department of Botany and Zoology Title of Thesis: The parasite fauna of economically important pelagic fishes in Lake Tanganyika Degree programme: Ecological and Evolutionary Biology Specialization: Parasitology Supervisor: Maarten Vanhove, Ph.D. Academic Year: 2019/2020 Number of Pages: 350 + 72 Keywords: Kapentagyrus, Dolicirrolectanum, Cryptogonimidae, Clupeidae, Latidae, Bathybatini Bibliografický záznam Autor: Mgr. Nikol Kmentová Přírodovědecká fakulta, Masarykova univerzita Ústav botaniky a zoologie Název práce: Parazité ekonomicky významných ryb pelagické zóny jezera Tanganika Studijní program: Ekologická a evoluční biologie Specializace: Parazitologie Vedoucí práce: Maarten Vanhove, Ph.D. Akademický rok: 2019/2020 Počet stran: 350 + 72 Klíčová slova: Kapentagyrus, Dolicirrolectanum, Cryptogonimidae, Clupeidae, Latidae, Bathybatini ABSTRACT Biodiversity is a well-known term characterising the variety and variability of life on Earth. It consists of many different levels with species richness as the most frequently used measure. Despite its generally lower species richness compared to littoral zones, the global importance of the pelagic realm in marine and freshwater ecosystems lies in the high level of productivity supporting fisheries worldwide. In terms of endemicity, Lake Tanganyika is one of the most exceptional freshwater study areas in the world. While dozens of studies focus on this lake’s cichlids as model organisms, our knowledge about the economically important fish species is still poor. Despite their important role in speciation processes, parasite taxa have been vastly ignored in the African Great Lakes including Lake Tanganyika for many years.