Fatal Neonatal-Onset Mitochondrial Respiratory Chain Disease with T Cell Immunodeficiency
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Proteomic and Metabolomic Analyses of Mitochondrial Complex I-Deficient
THE JOURNAL OF BIOLOGICAL CHEMISTRY VOL. 287, NO. 24, pp. 20652–20663, June 8, 2012 © 2012 by The American Society for Biochemistry and Molecular Biology, Inc. Published in the U.S.A. Proteomic and Metabolomic Analyses of Mitochondrial Complex I-deficient Mouse Model Generated by Spontaneous B2 Short Interspersed Nuclear Element (SINE) Insertion into NADH Dehydrogenase (Ubiquinone) Fe-S Protein 4 (Ndufs4) Gene*□S Received for publication, November 25, 2011, and in revised form, April 5, 2012 Published, JBC Papers in Press, April 25, 2012, DOI 10.1074/jbc.M111.327601 Dillon W. Leong,a1 Jasper C. Komen,b1 Chelsee A. Hewitt,a Estelle Arnaud,c Matthew McKenzie,d Belinda Phipson,e Melanie Bahlo,e,f Adrienne Laskowski,b Sarah A. Kinkel,a,g,h Gayle M. Davey,g William R. Heath,g Anne K. Voss,a,h René P. Zahedi,i James J. Pitt,j Roman Chrast,c Albert Sickmann,i,k Michael T. Ryan,l Gordon K. Smyth,e,f,h b2 a,h,m,n3 David R. Thorburn, and Hamish S. Scott Downloaded from From the aMolecular Medicine Division, gImmunology Division, and eBioinformatics Division, Walter and Eliza Hall Institute of Medical Research, Parkville, Victoria 3052, Australia, the bMurdoch Childrens Research Institute, Royal Children’s Hospital and Department of Paediatrics, University of Melbourne, Parkville, Victoria 3052, Australia, the cDépartement de Génétique Médicale, Université de Lausanne, 1005 Lausanne, Switzerland, the dCentre for Reproduction and Development, Monash Institute of Medical Research, Clayton, Victoria 3168, Australia, the hDepartment of Medical Biology -

Abstracts from the 50Th European Society of Human Genetics Conference: Electronic Posters
European Journal of Human Genetics (2019) 26:820–1023 https://doi.org/10.1038/s41431-018-0248-6 ABSTRACT Abstracts from the 50th European Society of Human Genetics Conference: Electronic Posters Copenhagen, Denmark, May 27–30, 2017 Published online: 1 October 2018 © European Society of Human Genetics 2018 The ESHG 2017 marks the 50th Anniversary of the first ESHG Conference which took place in Copenhagen in 1967. Additional information about the event may be found on the conference website: https://2017.eshg.org/ Sponsorship: Publication of this supplement is sponsored by the European Society of Human Genetics. All authors were asked to address any potential bias in their abstract and to declare any competing financial interests. These disclosures are listed at the end of each abstract. Contributions of up to EUR 10 000 (ten thousand euros, or equivalent value in kind) per year per company are considered "modest". Contributions above EUR 10 000 per year are considered "significant". 1234567890();,: 1234567890();,: E-P01 Reproductive Genetics/Prenatal and fetal echocardiography. The molecular karyotyping Genetics revealed a gain in 8p11.22-p23.1 region with a size of 27.2 Mb containing 122 OMIM gene and a loss in 8p23.1- E-P01.02 p23.3 region with a size of 6.8 Mb containing 15 OMIM Prenatal diagnosis in a case of 8p inverted gene. The findings were correlated with 8p inverted dupli- duplication deletion syndrome cation deletion syndrome. Conclusion: Our study empha- sizes the importance of using additional molecular O¨. Kırbıyık, K. M. Erdog˘an, O¨.O¨zer Kaya, B. O¨zyılmaz, cytogenetic methods in clinical follow-up of complex Y. -

A High-Fat Diet Coordinately Downregulates Genes Required for Mitochondrial Oxidative Phosphorylation in Skeletal Muscle Lauren M
A High-Fat Diet Coordinately Downregulates Genes Required for Mitochondrial Oxidative Phosphorylation in Skeletal Muscle Lauren M. Sparks, Hui Xie, Robert A. Koza, Randall Mynatt, Matthew W. Hulver, George A. Bray, and Steven R. Smith Obesity and type 2 diabetes have been associated with a ray studies have shown that genes involved in oxidative high-fat diet (HFD) and reduced mitochondrial mass phosphorylation (OXPHOS) exhibit reduced expression and function. We hypothesized a HFD may affect expres- levels in the skeletal muscle of type 2 diabetic subjects and sion of genes involved in mitochondrial function and prediabetic subjects. These changes may be mediated by biogenesis. To test this hypothesis, we fed 10 insulin- the peroxisome proliferator–activated receptor ␥ coacti- sensitive males an isoenergetic HFD for 3 days with vator-1 (PGC1) pathway. PGC1␣- and PGC1-responsive muscle biopsies before and after intervention. Oligonu- cleotide microarray analysis revealed 297 genes were OXPHOS genes show reduced expression in the muscle of differentially regulated by the HFD (Bonferonni ad- patients with type 2 diabetes (3,4). In addition to the justed P < 0.001). Six genes involved in oxidative cellular energy sensor AMP kinase, the peroxisome prolif- phosphorylation (OXPHOS) decreased. Four were mem- erator–activated receptor cofactors PGC1␣ (5–7) and pos- bers of mitochondrial complex I: NDUFB3, NDUFB5, sibly PGC1 (8) activate mitochondrial biogenesis and NDUFS1, and NDUFV1; one was SDHB in complex II and increase OXPHOS gene expression by increasing the tran- a mitochondrial carrier protein SLC25A12. Peroxisome scription, translation, and activation of the transcription proliferator–activated receptor ␥ coactivator-1 (PGC1) -and PGC1 mRNA were decreased by ؊20%, P < 0.01, factors necessary for mitochondrial DNA (mtDNA) repli ␣ and ؊25%, P < 0.01, respectively. -

Anti-NDUFS1 Polyclonal Antibody Cat: K110186P Summary
Anti-NDUFS1 Polyclonal Antibody Cat: K110186P Summary: 【Product name】: Anti-NDUFS1 Antibody 【Source】: Rabbit 【Isotype】: IgG 【Species reactivity】: Human Mouse Rat 【Swiss Prot】: P28331 【Gene ID】: 4719 【Calculated】: MW:68/73/75/79/81kDa 【Purification】: Octanoic acid-ammonium sulfate precipitation 【Tested applications】: IHC 【Recommended dilution】: IHC 1:50-200. 【IHC Positive sample】: Human breast cancer 【Subcellular location】: Cytoplasm 【Immunogen】: A synthetic peptide of human NDUFS1 【Storage】: Shipped at 4°C. Upon delivery aliquot and store at -20°C Background: The protein encoded by this gene belongs to the complex I 75 kDa subunit family. Mammalian complex I is composed of 45 different subunits. It locates at the mitochondrial inner membrane. This protein has NADH dehydrogenase activity and oxidoreductase activity. It transfers electrons from NADH to the respiratory chain. The immediate electron acceptor for the enzyme is believed to be ubiquinone. This protein is the largest subunit of complex I and it is a component of the iron-sulfur (IP) fragment of the enzyme. It may form part of the active site crevice where NADH is oxidized. Mutations in this gene are associated with complex I deficiency. Several transcript variants encoding different isoforms have been found for this gene. Sales:[email protected] Tech:[email protected] For research purposes only. Tel:400-968-6088 Please visit www.solarbio.com for a more product information Verified picture Immunohistochemistry of paraffin-embedded Human breast cancer with NDUFS1 antibody diluted at 1:100 Sales:[email protected] Tech:[email protected] For research purposes only. Tel:400-968-6088 Please visit www.solarbio.com for a more product information. -

Impaired Mitochondrial Fatty Acid Synthesis Leads to Neurodegeneration in Mice
This Accepted Manuscript has not been copyedited and formatted. The final version may differ from this version. Research Articles: Neurobiology of Disease Impaired mitochondrial fatty acid synthesis leads to neurodegeneration in mice Remya R. Nair1, Henna Koivisto2, Kimmo Jokivarsi2, Ilkka J. Miinalainen3, Kaija J. Autio1, Aki Manninen1,4, Heikki Tanila2, J. Kalervo Hiltunen1 and Alexander J. Kastaniotis1 1Faculty of Biochemistry and Molecular Medicine, University of Oulu, FI-90014 Oulu, Finland 2A. I. Virtanen Institute, University of Eastern Finland, FI-70211 Kuopio, Finland 3Electron Microscopy Core Facility, Biocenter Oulu, University of Oulu, FI-90014 Oulu, Finland 4Virus Core Facility, Biocenter Oulu, University of Oulu, FI-90014 Oulu, Finland DOI: 10.1523/JNEUROSCI.3514-17.2018 Received: 13 December 2017 Revised: 31 August 2018 Accepted: 19 September 2018 Published: 28 September 2018 Author contributions: R.R.N., A.M., H.T., J.K.H., and A.J.K. designed research; R.R.N., H.K., K.J., I.M., K.J.A., H.T., and J.K.H. performed research; R.R.N., H.K., K.J., I.M., K.J.A., A.M., H.T., J.K.H., and A.J.K. analyzed data; R.R.N., H.K., K.J., I.M., K.J.A., A.M., H.T., J.K.H., and A.J.K. wrote the paper; A.M. contributed unpublished reagents/analytic tools. Conflict of Interest: The authors declare no competing financial interests. We are grateful to Dr Raija Soininen for the continuous support at various stages of this project and critical review of this manuscript. The Biocenter Oulu Transgenic Core facility, the Biocenter Oulu EM laboratory and the Biocenter Oulu Virus Core laboratory, which are all part of Biocenter Finland, are acknowledged for the support in generating mice, in the EM analysis and the lentivirus production, respectively. -

Inositol Phosphate in the Basidiomycete Fungus Schizophyllum Commune
Inositol phosphate in the basidiomycete fungus Schizophyllum commune Dissertation To fulfill the Requirements for the Degree of „doctor rerum naturalium“ (Dr. rer. nat.) Submitted to the Council of the Faculty of Biological Science of the Friedrich Schiller University Jena by Reyna Carmina Felicia Murry, MSc. born on the 22nd of October 1987 in Bandung, Indonesia First reviewer: Prof. Dr. Erika Kothe, Institut für Mikrobiologie, FSU Jena Second Reviewer: Prof. Dr. Axel A. Brakhage, Institut für Mikrobiologie, FSU Jena Third Reviewer: Prof. Dr. J. Stephen Horton, Department of Biological Sciences, Science and Engineering Center, Union College, Schenectady, New York, USA Date of public defense: 7th May 2019 “Science is the key to our future, and if you don't believe in science, then you're holding everybody back.” Bill Nye Table of Contents 1 Introduction ......................................................................................................................... 1 1.1 Schizophyllum commune .............................................................................................. 1 1.2 Fruiting body development in S. commune ................................................................. 3 1.3 Signal transduction: G-protein-coupled receptors (GPCRs) and Ras signaling .......... 4 1.4 Inositol-based metabolism and signaling: inositol phosphates and inositol lipids ...... 5 1.5 Inositol and inositol monophosphatase ........................................................................ 7 1.6 Inositol hexakisphosphate and -

A Novel Mutation in NDUFS4 Causes Leigh Syndrome in an Ashkenazi Jewish Family
J Inherit Metab Dis (2008) 31 (Suppl 2):S461–S467 DOI 10.1007/s10545-008-1049-9 SHORT REPORT A novel mutation in NDUFS4 causes Leigh syndrome in an Ashkenazi Jewish family S. L. Anderson & W. K. Chung & J. Frezzo & J. C. Papp & J. Ekstein & S. DiMauro & B. Y. Rubin Received: 15 September 2008 /Submitted in revised form: 13 November 2008 /Accepted: 19 November 2008 / Published online: 26 December 2008 # SSIEM and Springer 2008 Summary Leigh syndrome is a neurodegenerative without consanguinity with three affected children. disorder of infancy or childhood generally due to Linkage to microsatellite markers D5S1969 and mutations in nuclear or mitochondrial genes involved D5S407 led to evaluation of the complex I gene in mitochondrial energy metabolism. We performed NDUFS4, in which we identified a novel homozygous linkage analysis in an Ashkenazi Jewish (AJ) family c.462delA mutation that disrupts the reading frame. The resulting protein lacks a cAMP-dependent protein kinase phosphorylation site required for activation of Communicating editor: John Christodoulou mitochondrial respiratory chain complex I. In a Competing interests: None declared random sample of 5000 healthy AJ individuals, the References to electronic databases: Leigh syndrome: OMIM carrier frequency of the NDUFS4 mutation c.462delA 256000. NDUFS4: OMIM 602694. NDUFS4 mRNA: GenBank was 1 in 1000, suggesting that it should be considered accession # NM_002495. : : in all AJ patients with Leigh syndrome. S. L. Anderson J. Frezzo B. Y. Rubin (*) Department of Biological Sciences, Fordham University, Abbreviations 441 E. Fordham Rd., Bronx, NY 10458, USA AJ Ashkenazi Jewish e-mail: [email protected] BCS1L BCS1-like protein W. -
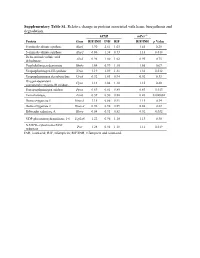
Supplementary Table S1. Relative Change in Proteins Associated with Heme Biosynthesis and Degradation
Supplementary Table S1. Relative change in proteins associated with heme biosynthesis and degradation. hPXR mPxr–/– Protein Gene RIF/INH INH RIF RIF/INH p Value 5-aminolevulinate synthase Alas1 1.90 2.61 1.05 1.41 0.28 5-aminolevulinate synthase Alas2 0.86 1.38 0.73 1.18 0.018 Delta-aminolevulinic acid Alad 0.96 1.00 1.02 0.95 0.75 dehydratase Porphobilinogen deaminase Hmbs 1.04 0.99 1.10 1.05 0.67 Uroporphyrinogen-III synthase Uros 1.19 1.09 1.31 1.38 0.012 Uroporphyrinogen decarboxylase Urod 0.92 1.03 0.94 0.92 0.33 Oxygen-dependent Cpox 1.13 1.04 1.18 1.15 0.20 coproporphyrinogen-III oxidase, Protoporphyrinogen oxidase Ppox 0.69 0.81 0.85 0.83 0.013 Ferrochelatase, Fech 0.39 0.50 0.88 0.43 0.000002 Heme oxygenase 1 Hmox1 1.15 0.86 0.91 1.11 0.34 Heme oxygenase 2 Hmox2 0.96 0.98 0.89 0.88 0.22 Biliverdin reductase A Blvra 0.84 0.92 0.82 0.92 0.032 UDP-glucuronosyltransferase 1-6 Ugt1a6 1.22 0.96 1.10 1.13 0.30 NADPH--cytochrome P450 Por 1.28 0.92 1.18 1.12 0.019 reductase INH, isoniazid; RIF, rifampicin; RIF/INH, rifampicin and isoniazid. Supplementary Table S2. Relative change in protein nuclear receptors. hPXR mPxr–/– Protein Gene RIF/INH INH RIF RIF/INH p Value Aryl hydrocarbon receptor Ahr 1.09 0.91 1.00 1.26 0.092 Hepatocyte nuclear factor Hnf1a 0.87 0.97 0.82 0.79 0.027 1-alpha Hepatocyte nuclear factor Hnf4a 0.95 1.05 0.97 1.08 0.20 4-alpha Oxysterols receptor LXR- Nr1h3 0.94 1.16 1.03 1.02 0.42 alpha Bile acid receptor Nr1h4 1.05 1.17 0.98 1.19 0.12 Retinoic acid receptor Rxra 0.88 1.03 0.83 0.95 0.12 RXR-alpha Peroxisome proliferator- -

Epigenetic Programming by Prenatal Stress in Female Serotonin Transporter Deficient Mice
Epigenetic programming by prenatal stress in female serotonin transporter deficient mice Epigenetische Programmierung durch Pränatalstress in weiblichen Serotonintransporter-defizienten Mäusen Doctoral thesis for a doctoral degree at the Graduate School of Life Sciences, Julius-Maximilians-Universität Würzburg, Section Neuroscience Submitted by Karla-Gerlinde Schraut from Timişoara, România Würzburg, 2015 Submitted on: ............................................................................. Members of the Promotionskomitee: Chairperson: Prof. Dr. Thomas Dandekar Primary Supervisor: Prof. Dr. Klaus-Peter Lesch Supervisor (Second): Prof. Dr. Charlotte Förster Supervisor (Third): Dr. Daniel van den Hove Date of Public Defense: ............................................................... Date of Receipt of Certificates: .................................................... Table of Contents Table of Contents ................................................................................................................................ I Abbreviations ..................................................................................................................................... V Summary…………………………………………………………………………………………………..XI Zusammenfassung ........................................................................................................................... XV 1. General introduction ................................................................................................... 1 1.1. Variation in the serotonin transporter -

Analysis of Human Mutations in the Supernumerary Subunits of Complex I
life Review Analysis of Human Mutations in the Supernumerary Subunits of Complex I Quynh-Chi L. Dang, Duong H. Phan, Abigail N. Johnson, Mukund Pasapuleti, Hind A. Alkhaldi, Fang Zhang and Steven B. Vik * Department of Biological Sciences, Southern Methodist University, Dallas, TX 75287, USA; [email protected] (Q.-C.L.D.); [email protected] (D.H.P.); [email protected] (A.N.J.); [email protected] (M.P.); [email protected] (H.A.A.); [email protected] (F.Z.) * Correspondence: [email protected] Received: 30 October 2020; Accepted: 16 November 2020; Published: 20 November 2020 Abstract: Complex I is the largest member of the electron transport chain in human mitochondria. It comprises 45 subunits and requires at least 15 assembly factors. The subunits can be divided into 14 “core” subunits that carry out oxidation–reduction reactions and proton translocation, as well as 31 additional supernumerary (or accessory) subunits whose functions are less well known. Diminished levels of complex I activity are seen in many mitochondrial disease states. This review seeks to tabulate mutations in the supernumerary subunits of humans that appear to cause disease. Mutations in 20 of the supernumerary subunits have been identified. The mutations were analyzed in light of the tertiary and quaternary structure of human complex I (PDB id = 5xtd). Mutations were found that might disrupt the folding of that subunit or that would weaken binding to another subunit. In some cases, it appeared that no protein was made or, at least, could not be detected. A very common outcome is the lack of assembly of complex I when supernumerary subunits are mutated or missing. -

PRAVEEN KUMAR DHANDAPANI: Expressing Alternative Oxidase
PRAVEEN KUMAR DHANDAPANI DHANDAPANI KUMAR PRAVEEN DISSERTATIONES SCHOLAE DOCTORALIS AD SANITATEM INVESTIGANDAM EXPRESSING ALTERNATIVE OXIDASE (AOX) IN MICE: A VALUABLE TOOL TO STUDY MITOCHONDRIAL RESPIRATORY CHAIN DEFECTS RESPIRATORY MITOCHONDRIAL STUDY TO TOOL IN MICE: A VALUABLE (AOX) OXIDASE ALTERNATIVE EXPRESSING UNIVERSITATIS HELSINKIENSIS PRAVEEN KUMAR DHANDAPANI EXPRESSING ALTERNATIVE OXIDASE (AOX) IN MICE: A VALUABLE TOOL TO STUDY MITOCHONDRIAL RESPIRATORY CHAIN DEFECTS ISBN 978-951-51-5998-4 (PRINT) ISBN 978-951-51-5999-1 (ONLINE) ISSN 2342-3161 (PRINT) ISSN 2342-317X (ONLINE) http://ethesis.helsinki.fi HELSINKI 2020 FACULTY OF MEDICINE DOCTORAL PROGRAM IN BIOMEDICINE UNIVERSITY OF HELSINKI 32/2020 Doctoral Programme in Biomedicine Faculty of Medicine University of Helsinki Finland Expressing alternative oxidase (AOX) in mice: A valuable tool to study mitochondrial respiratory chain defects Praveen Kumar Dhandapani ACADEMIC DISSERTATION To be presented, with the permission of Faculty of Medicine of the University of Helsinki, for public examination in room P674, Porthania, Yliopistonkatu 3, Helsinki, on 05.06.2020, at noon. Helsinki 2020 Supervised by Marten Szibor, MD Prof. Howard T. Jacobs, PhD Clinic for Heart and Thorax Surgery Faculty of Medicine and Health Technology Jena University Hospital Tampere University Friedrich-Schiller University of Jena Thesis committee Prof. Katriina Aalto-Setälä, MD, PhD Prof. Klaus-Dieter Schlüter, PhD Faculty of Medicine and Health Physiological Institute Technology Faculty of Medicine Tampere University Justus-Liebig University of Giessen Reviewed by Jaakko Pohjoismäki, PhD Prof. Roberta A. Gottlieb, MD Department of Environmental and Department of Medicine; Department of Biological Sciences Biomedical Sciences University of Eastern Finland Smidt Heart Institute Cedars-Sinai Medical Center Opponent Prof. -

Activation of the Endogenous Renin-Angiotensin- Aldosterone System Or Aldosterone Administration Increases Urinary Exosomal Sodium Channel Excretion
CLINICAL RESEARCH www.jasn.org Activation of the Endogenous Renin-Angiotensin- Aldosterone System or Aldosterone Administration Increases Urinary Exosomal Sodium Channel Excretion † † Ying Qi,* Xiaojing Wang, Kristie L. Rose,* W. Hayes MacDonald,* Bing Zhang, ‡ | Kevin L. Schey,* and James M. Luther § Departments of *Biochemistry, †Bioinformatics, ‡Division of Clinical Pharmacology, Department of Medicine, §Division of Nephrology, Department of Medicine, and |Department of Pharmacology, Vanderbilt University School of Medicine, Nashville, Tennessee ABSTRACT Urinary exosomes secreted by multiple cell types in the kidney may participate in intercellular signaling and provide an enriched source of kidney-specific proteins for biomarker discovery. Factors that alter the exosomal protein content remain unknown. To determine whether endogenous and exogenous hormones modify urinary exosomal protein content, we analyzed samples from 14 mildly hypertensive patients in a crossover study during a high-sodium (HS, 160 mmol/d) diet and low-sodium (LS, 20 mmol/d) diet to activate the endogenous renin-angiotensin-aldosterone system. We further analyzed selected exosomal protein content in a separate cohort of healthy persons receiving intravenous aldosterone (0.7 mg/kg per hour for 10 hours) versus vehicle infusion. The LS diet increased plasma renin activity and aldosterone concentration, whereas aldosterone infusion increased only aldosterone concentration. Protein analysis of paired urine exosome samples by liquid chromatography-tandem mass spectrometry–based multidimen- sional protein identification technology detected 2775 unique proteins, of which 316 exhibited signifi- cantly altered abundance during LS diet. Sodium chloride cotransporter (NCC) and a-andg-epithelial sodium channel (ENaC) subunits from the discovery set were verified using targeted multiple reaction monitoring mass spectrometry quantified with isotope-labeled peptide standards.