Genetics of Amyotrophic Lateral Sclerosis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Fibromyalgia and Chronic Fatigue
Fibromyalgia and Chronic Fatigue Paul G. Swingle, Ph.D., R. Psych. My previous radio program and my present webcast program are both entitled “It’s all in your head!” How I came up with this title was listening to the discouraged patients that I routinely treated, and still treat, who have been told, by the doctors they have consulted, that the pain, discomfort, distress, debilitating fatigue, sleep disturbance and depression that they endure is “all in your head.” What this remark is meant to imply, of course, is that there is no physical cause of the condition and that the symptoms are manifestations of psychological factors. My response to these patients is that “of course it is in your head, where else would it be?” My remark however is not disparaging but rather indicates the actual location of many of the patients symptoms. The brain controls everything so all symptoms are associated with brain activity. The remarkable effectiveness of neurotherapy for the treatment of the symptoms associated with fibromyalgia and chronic fatigue is due to the fact that the brain functioning associated with the symptoms is treated. Although this derogatory sentiment about fibromyalgia patients was widespread among health care providers in the past, it is remarkable to me that it still persists with many doctors to this day. A few days ago I was surfing the internet health news feeds and was dismayed to find an item entitled “Fibromyalgia: A mental illness?” In this item was a quote from a Canadian neurologist stating that fibromyalgia was a term waiting for a definition. -

Vascular Dementia: an Overview of Current Challenges and Gaps
Stroke and Dementia: what research tells us UK Stroke Assembly 2016 JM Wardlaw University of Edinburgh NHS Lothian www.strokeassembly.org.uk #strokeassembly Definitions Stroke Dementia Sudden onset focal A syndrome where there neurological deficit which is deterioration in last more than 24 hours memory, thinking, attributable to a vascular behaviour such that the defect and has no other person is no longer able identifiable cause to perform everyday activities by themselves www.strokeassembly.org.uk #strokeassembly Stroke – the importance • Stroke, 16.9million new strokes per year, – A third are disabled and dependent – 2nd commonest cause of death – Commonest cause of dependency in adults Infarct, or ischaemic stroke Haemorrhagic stroke www.strokeassembly.org.uk #strokeassembly Cerebrovascular disease – the importance • ‘Silent’ cerebrovascular disease, even bigger issue – Up to 45% of 35.6million dementias – Cognitive impairment, mobility problems Normal White matter disease www.strokeassembly.org.uk #strokeassembly Dementia • 47.5 million people have dementia world wide • 7.7million new cases per year • Affects different people in different ways • Major cause of dependency and disability amongst older people • Alzheimer’s disease commonest cause – 60-70% of cases • Vascular dementia 2nd commonest – 25-45% of all cases www.strokeassembly.org.uk #strokeassembly Dementia: historical perspective 1800’s – Blood vessel diseases 1900’s – Alzheimers disease - dominant 1970’s – ‘Multi-infarct dementia’ – vascular 1990’s–2000’s - ‘Vascular -

TIA Vs CVA (STROKE)
Phone: 973.334.3443 Email: [email protected] NJPR.com TIA vs CVA (STROKE) What is the difference between a TIA and a stroke? Difference Between TIA and Stroke • Both TIA and stroke are due to poor blood supply to the brain. • Stroke is a medical emergency and it’s a life-threatening condition. • The symptoms of TIA and Stroke may be same but TIA symptoms will recover within 24 hours. TRANSIENT ISCHEMIC ATTACK ● Also known as: TIA, mini stroke 80 E. Ridgewood Avenue, 4th Floor Paramus, NJ 07652 TIA Causes ● A transient ischemic attack has the same origins as that of an ischemic stroke, the most common type of stroke. In an ischemic stroke, a clot blocks the blood supply to part of your brain. In a transient ischemic attack, unlike a stroke, the blockage is brief, and there is no permanent damage. ● The underlying cause of a TIA often is a buildup of cholesterol- containing fatty deposits called plaques (atherosclerosis) in an artery or one of its branches that supplies oxygen and nutrients to your brain. ● Plaques can decrease the blood flow through an artery or lead to the development of a clot. A blood clot moving to an artery that supplies your brain from another part of your body, most commonly from your heart, also may cause a TIA. CEREBROVASCULAR ACCIDENT/STROKE Page 2 When the brain’s blood supply is insufficient, a stroke occurs. Stroke symptoms (for example, slurring of speech or loss of function in an arm or leg) indicate a medical emergency. Without treatment, the brain cells quickly become impaired or die. -
ICD-10 Backs
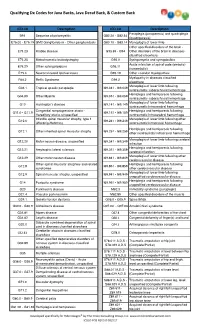
Qualifying Dx Codes for Java Backs, Java Decaf Back, & Custom Back ICD-10 Description ICD-10 Description Paraplegia (paraparesis) and quadriplegia B91 Sequelae of poliomyelitis G82.20 - G82.54 (quadriparesis) E75.00 - E75.19 GM2 Gangliosidosis - Other gangliosidosis G83.10 - G83.14 Monoplegia of lower limb Other specified disorders of the brain - E75.23 Krabbe disease G93.89 - G94 Other disorders of the brain in diseases classified elsewhere E75.25 Metachromatic leukodystrophy G95.0 Syringomyelia and syringobulbia Acute infarction of spinal code (embolic) E75.29 Other sphingolipidosis G95.11 (nonembolic) E75.4 Neuronal ceroid lipofuscinosis G95.19 Other vascular myelopathies Myelopathy in diseases classified F84.2 Rett's Syndrome G99.2 elsewhere Monoplegia of lower limb following G04.1 Tropical spastic paraplegia I69.041 - I69.049 nontraumatic subarachnoid hemorrhage Hemiplegia and hemiparesis following G04.89 Other Myelitis I69.051 - I69.059 nontraumatic subarachnoid hemorrhage Monoplegia of lower limb following G10 Huntington's disease I69.141 - I69.149 nontraumatic intracerebral hemorrhage Congenital nonprogressive ataxia - Hemiplegia and hemiparesis following G11.0 - G11.9 I69.151 - I69.159 Hereditary ataxia, unspecified nontraumatic intracerebral hemorrhage Infantile spinal muscular atrophy, type 1 Monoplegia of lower limb following other G12.0 I69.241 - I69.249 (Werdnig-Hoffman) nontraumatic intracranial hemorrhage Hemiplegia and hemiparesis following G12.1 Other inherited spinal muscular atrophy I69.251 - I69.259 other nontraumatic -

Central Pain: Clinical and Physiological J Neurol Neurosurg Psychiatry: First Published As 10.1136/Jnnp.61.1.62 on 1 July 1996
626Journal ofNeurology, Neurosurgery, and Psychiatry 1996;61:62-69 Central pain: clinical and physiological J Neurol Neurosurg Psychiatry: first published as 10.1136/jnnp.61.1.62 on 1 July 1996. Downloaded from characteristics David Bowsher Abstract spinothalamic pathway, its relays, or projec- Objectives-To study the clinical and tions may cause central pain,' and modern pathophysiological features of central radiological techniques have tended to con- pain due to damage to the CNS. firm this.9 '" Because of this, and of recent Methods-156 patients (mostly with considerations on pathophysiology,'5 '" the ischaemic strokes, some with infarct after condition is now known as "central poststroke subarachnoid haemorrhage and other pain (CPSP)"'.'7 Stroke is not the only condi- cerebral conditions; one with bulbar and tion causing such central pains of cerebral ori- others with spinal pathology) with central gin.' lxII 2' Furthermore, identical pains occur pain have been investigated clinically and after some spinal lesions.- 22 varying numbers instrumentally with respect to quantitative somatosensory perception thresholds and autonomic Patients function. Between 1983 and 1993, 156 patients have Results-Pain onset was immediate in a been referred to me, in whom a diagnosis of minority; and from a week or two up to central pain due to a cerebral or spinal lesion six years in > 60%. For those with supra- has been made. Of these, 112 (64 (57)%'Y spinal ischaemic lesions, the median age male) had a history of a stroke episode (cere- of onset was 59; dominant and non- brovascular accident, CVA patients), includ- dominant sides were equally affected. -

Transient Ischemic Attacks (Tias) and Strokes (Cvas)
CREATED EXCLUSIVELY FOR FINANCIAL PROFESSIONALS Transient Ischemic Attacks (TIAs) and Rx FOR SUCCESS Strokes (CVAs) Obstruction in blood flow (ischemia) to the brain can lead to permanent damage. This is called a cerebrovascular accident (CVA). It is also known as cerebral infarction or stroke. If the symptoms are temporary without permanent brain damage, the event is called a transient ischemic attack (TIA). Rupture of an artery with bleeding into the brain (hemorrhage) is called a CVA, too. Strokes and TIAs are rated based on the underlying cause. The most common cause of TIAs and CVAs is hypertensive and atherosclerotic plaque within the arteries to the brain (aka cerebrovascular disease or CVD). CVD can be complicated by clots (thrombosis) and by emboli from the heart. Because CVD is an indicator of atherosclerosis in other parts of the body, an individual with a history of TIA or CVA is at risk for coronary artery disease and recurrent stroke. Risk factors for CVD include smoking, coronary artery disease, high blood pressure, diabetes, lipid disorders (such as high cholesterol), peripheral arterial disease, and atrial fibrillation. Signs and symptoms of a CVA/TIA include weakness, numbness, headaches, dizziness, nausea, vomiting, paralysis of one side of the body, speech difficulty, and memory defects. Amaurosis fugax, a form of visual TIA, is temporary monocular (one eye) or partial blindness. Tests are done to evaluate the brain circulation, such as a carotid ultrasound (Duplex) or angiogram (MRA). A brain scan (CT and/or MRI) is used to determine if an individual has had a stroke. A TIA will not show on a scan. -

Pathophysiology and Treatment of Stroke: Present Status and Future Perspectives
International Journal of Molecular Sciences Review Pathophysiology and Treatment of Stroke: Present Status and Future Perspectives Diji Kuriakose and Zhicheng Xiao * Development and Stem Cells Program, Monash Biomedicine Discovery Institute and Department of Anatomy and Developmental Biology, Monash University, Melbourne, VIC 3800, Australia; [email protected] * Correspondence: [email protected] Received: 29 September 2020; Accepted: 13 October 2020; Published: 15 October 2020 Abstract: Stroke is the second leading cause of death and a major contributor to disability worldwide. The prevalence of stroke is highest in developing countries, with ischemic stroke being the most common type. Considerable progress has been made in our understanding of the pathophysiology of stroke and the underlying mechanisms leading to ischemic insult. Stroke therapy primarily focuses on restoring blood flow to the brain and treating stroke-induced neurological damage. Lack of success in recent clinical trials has led to significant refinement of animal models, focus-driven study design and use of new technologies in stroke research. Simultaneously, despite progress in stroke management, post-stroke care exerts a substantial impact on families, the healthcare system and the economy. Improvements in pre-clinical and clinical care are likely to underpin successful stroke treatment, recovery, rehabilitation and prevention. In this review, we focus on the pathophysiology of stroke, major advances in the identification of therapeutic targets and recent trends in stroke research. Keywords: stroke; pathophysiology; treatment; neurological deficit; recovery; rehabilitation 1. Introduction Stroke is a neurological disorder characterized by blockage of blood vessels. Clots form in the brain and interrupt blood flow, clogging arteries and causing blood vessels to break, leading to bleeding. -

Turn Off the Amyotrophic Lateral Sclerosis's Disease Gene
Meet the Professor Page 1 of 4 Prof. Dongsheng Fan: turn off the amyotrophic lateral sclerosis’s disease gene Submitted Nov 11, 2014. Accepted for publication Nov 12, 2014. doi: 10.3978/j.issn.2305-5839.2014.11.14 View this article at: http://dx.doi.org/10.3978/j.issn.2305-5839.2014.11.14 With more and more celebrities all over the world jumping on the bandwagon of the Ice Bucket Challenge, amyotrophic lateral sclerosis (ALS) has been put in the spotlight. Though this event in a way had risen the awareness of ALS, there are still so many unsolved problems about it, such as the difficulty of accurate diagnosis and complete treatment. To discuss the hotspots and recent advances in this area, Annals of Translational Medicine has the pleasure to invite Prof. Dongsheng Fan (Figure 1), one of the leading experts on degenerative diseases of the nervous system in China, to unveil the “mystery” of ALS and find out what’s newest about this disease. Prof. Fan will also work on a special issue about ALS as the guest editor of Annals of Translational Medicine subsequently. Figure 1 Professor Dongsheng Fan. Introduction Professor Dongsheng Fan is currently the Chief of Neurology in the Peking University Third Hospital, From a professional perspective, what are the disease a member of the standing committee of the Chinese characteristics of ALS? Neurological Society, the vice-chairman of the Beijing Prof. Fan: Clinically, ALS, AD, PD, and multiple system Branch of the Chinese Academy of Neurology, a member atrophy (MSA) are all typical degenerative diseases of the of the Board of Directors of the World Stroke Organization nervous system. -

Guidelines for Diagnosis and Treatment of Moyamoya Disease (Spontaneous Occlusion of the Circle of Willis)
Neurol Med Chir (Tokyo) 52, 245¿266, 2012 Guidelines for Diagnosis and Treatment of Moyamoya Disease (Spontaneous Occlusion of the Circle of Willis) Research Committee on the Pathology and Treatment of Spontaneous Occlusion of the Circle of Willis; Health Labour Sciences Research Grant for Research on Measures for Intractable Diseases CHAPTER I: CONCEPTS OF THE DISEASE Concepts of the Disease (ii) Abnormal vascular networks in the vicinity of the occlusive or stenotic lesions in the ar- The characteristics of moyamoya disease (spontane- terial phase. ous occlusion of the circle of Willis, cerebrovascular (iii) Bilaterality of findings (i) and (ii). ``moyamoya'' disease) on cerebral angiography were (2) However, when magnetic resonance imaging reported for the first time in 1957,8) and the concept (MRI) and magnetic resonance angiographic of moyamoya disease as a separate disease entity (MRA) findings meet all of the following criteria, was established in the 1960s.1–3,6,7) Pathologically, cerebral angiography can be omitted. See the moyamoya disease is characterized by chronic ``Guidelines for Diagnostic Imaging by MRI and progressive stenosis of the terminal portion of the MRA.'' bilateral internal carotid arteries, which leads to the (i) MRA shows stenosis or occlusion of the ter- formation of an abnormal vascular network com- minal portion of the intracranial internal posed of collateral pathways at the base of the brain carotid artery or proximal portions of the (moyamoya vessels at the base of the brain) anterior and/or the middle cerebral artery. (`moyamoya' is the Japanese term for a ``puff of (ii) MRA shows abnormal vascular networks in smoke,'' which has been used to describe the appear- the basal ganglia. -

CNS Findings in Chronic Fatigue Syndrome and a Neuropathological Case Report Kimberly Ferrero, Mitchell Silver, Alan Cocchetto, Eliezer Masliah and Dianne Langford
Downloaded from http://jim.bmj.com/ on April 7, 2017 - Published by group.bmj.com Original research CNS findings in chronic fatigue syndrome and a neuropathological case report Kimberly Ferrero,1 Mitchell Silver,1 Alan Cocchetto,2 Eliezer Masliah,3 Dianne Langford1 1Lewis Katz School of ABSTRACT Medicine at Temple Chronic fatigue syndrome (CFS) is characterized as a Significance of this study University, Philadelphia, Pennsylvania, USA persistent, debilitating complex disorder of unknown 2State University of etiology, whereby patients suffer from extreme New York at Alfred, fatigue, which often presents with symptoms that What is already known about this Engineering Technologies, include chronic pain, depression, weakness, mood subject? Alfred, New York, USA disturbances, and neuropsychological impairment. In ▸ 3University of California San Chronic fatigue syndrome (CFS) is 6 months Diego, La Jolla, California, this mini review and case report, we address central or more of persistent cognitive and physical USA nervous system (CNS) involvement of CFS and fatigue, with brain-related symptoms present neuropathological autopsy findings from a varying among patients Correspondence to patient who died with a prior diagnosis of CFS. ▸ CFS is classified as a neurological disorder Dianne Langford, Lewis Katz School of Medicine at Among the most remarkable pathological features of and increasing evidence supports CFS as a Temple University, the case are focal areas of white matter loss, neurite disease of the nervous and immune 3500 North Broad Street, beading, and neuritic pathology of axons in the systems MERB 750, Philadelphia, white matter with axonal spheroids. Atypical ▸ In the central nervous system (CNS), CFS PA 19140, USA; neurons displaying aberrant sprouting processes in [email protected] may be triggered by exposure to response to injury are observed throughout cortical radionuclides, viral or microbial infection, Accepted 14 March 2017 gray and white matter. -

Contents Editorial Commentaries Multiple Sclerosis
March 2019 Volume 90 Issue 3 90 Impact 3 Factor 7.144 Volume 90 Issue 3 Volume Contents Volume 90 Issue 3 | JNNP March 2019 Pages 247–368 Pages JOURNAL OF NEUROLOGY NEUROSURGERY & PSYCHIATRY NEUROSURGERY OF NEUROLOGY JOURNAL Editorial commentaries Neuromuscular J Neurol Neurosurg Psychiatry: first published as on 1 March 2019. Downloaded from ISSUE HIGHLIGHTS 247 The unintended consequences of NICE 294 Neurochemical correlates of functional decline Epilepsy – Surgery at it’s best Amyotrophic Lateral Sclerosis – Age of onset G Giovannoni Multiple Sclerosis – 10 years of risk sharing in amyotrophic lateral sclerosis Stroke – Endovascular therapy outcomes Neuropathy – Mining the genes PTSD – Therapy for Veterans I Cheong, D K Deelchand, L E Eberly, M Marja ska, G Manousakis, G Guliani, D Walk, March 2019 March 249 Epilepsy surgery at its best: randomised ń jnnp.bmj.com prospective controlled trials in neurosurgery G Öz Cover image: Brain tree. are no magic Editor P C Warnke Cognitive neurology Matthew C Kiernan (Australia) 302 Brain mechanisms underlying apathy Deputy Editors C Le Heron, C B Holroyd, J Salamone, M Husain Karen L Furie (USA) 250 What does age at onset in ALS tell us about Satoshi Kuwabara (Japan) the genetic basis of the disease? Lawrence Wong (China) J H Veldink Associate Editors Cerebrovascular disease Alan Carson (UK) 313 Impact of infarct location on functional Nick Ward (UK) Multiple sclerosis outcome following endovascular therapy for Peter Warnke (USA) 251 Assessing the long-term effectiveness of Commissioning Editor -

Dissecting the Association Between Migraine and Stroke
Curr Neurol Neurosci Rep (2015) 15:5 DOI 10.1007/s11910-015-0530-8 HEADACHE (RB HALKER, SECTION EDITOR) Dissecting the Association Between Migraine and Stroke Andrea M. Harriott & Kevin M. Barrett # Springer Science+Business Media New York 2015 Abstract Migraine is a common disabling neurological dis- Introduction order resulting from excessive cortical excitation and trigeminovascular afferent sensitization. In addition to aber- Migraine is a common neurological condition characterized rant neuronal processing, migraineurs are also at significant by recurrent unilateral headaches of moderate to severe inten- risk of vascular disease. Consequently, the impact of mi- sity lasting 4–72 h in duration. The pain is often pulsatile in graine extends well beyond the ictal headache and includes quality and associated with nausea, vomiting, and sensitivity a well-documented association with acute ischemic stroke, to light and sound. Migraines can be heralded by visual aura in particularly in young women with a history of migraine with addition to other somatosensory, vestibular, motor, and aura. The association between migraine and stroke has been speech-related disturbances [1–4]. Migraine impacts 15 to acknowledged for 40 years or more. However, examining 20 % of the general adult population, including approximately the pathobiology of this association has become a more 17 to 25 % of women and 6 to 10% of men [5–7]. Starting as recent and critically important undertaking. The diversity early as 12 years of age, the highest prevalence of migraine is of mechanisms underlying the association between migraine amongst persons 20 to 50 years of age [6, 8]. The social, and stroke likely reflects the heterogenous nature of this economic, and psychological burden of migraine alone can disorder.