Resolving the Evolutionary Relationships of Molluscs with Phylogenomic Tools
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

27-60, 1999 Redescription Problematic Alpine Oxychilus
BASTERIA, 63: 27-60, 1999 Redescription of two problematic Alpine Oxychilus: O. adamii (Westerlund, 1886) and O. polygyra (Pollonera, 1885) (Pulmonata, Zonitidae) F. Giusti & G. Manganelli Dipartimento di Biologia Evolutiva dell'Universita di Siena, Via P.A. Mattioli 4, 1-53100 Siena, Italy of The taxonomic and nomenclatural status Oxychilus adamii (Westerlund, 1886) and O. polygyra is (Pollonera, 1885) revised. These two, very similar species are tentatively assigned to Mediterranean of characterized absent short a ‘subgenus’ Oxychilus by (1) or very flagellum; (2) penial retractor inserted where epiphallus ends and proximal penis begins; (3) internal ornamentation of penis of and of of consisting pleats rows papillae, some which or all with apical thorn; (4) short slender epiphallus internally with series of transverse crests on one side and a few longitudinal the pleats on other; (5) mucous gland forming muff of glandular tissue, denser and yellower around distal portion of free oviduct; (6) and short mesocone on the central tooth. O. adamii is medium-sized shell 16.1 ± 1.0 distinguished by a (diameter mm) and very small papillae withoutapical thorn covering the initialportion of the proximal penis. O. polygyra is distinguished small shell 10.4 ± 0.5 and by a (diameter mm) large papillae with an evident cuticularized The with apical thorn covering the initial portion of the proximal penis. paper concludes a review of the current status of the Oxychilus taxonomy, including main problems and some tentative solutions to be verified in the context of individual revisions. words: Key Gastropoda, Pulmonata, Zonitidae, Oxychilus, taxonomy, nomenclature, Italy. INTRODUCTION Ten species of Oxychilus Fitzinger, 1833, are reported from the Alps: O. -

Fauna of New Zealand Ko Te Aitanga Pepeke O Aotearoa
aua o ew eaa Ko te Aiaga eeke o Aoeaoa IEEAE SYSEMAICS AISOY GOU EESEAIES O ACAE ESEAC ema acae eseac ico Agicuue & Sciece Cee P O o 9 ico ew eaa K Cosy a M-C aiièe acae eseac Mou Ae eseac Cee iae ag 917 Aucka ew eaa EESEAIE O UIESIIES M Emeso eame o Eomoogy & Aima Ecoogy PO o ico Uiesiy ew eaa EESEAIE O MUSEUMS M ama aua Eiome eame Museum o ew eaa e aa ogaewa O o 7 Weigo ew eaa EESEAIE O OESEAS ISIUIOS awece CSIO iisio o Eomoogy GO o 17 Caea Ciy AC 1 Ausaia SEIES EIO AUA O EW EAA M C ua (ecease ue 199 acae eseac Mou Ae eseac Cee iae ag 917 Aucka ew eaa Fauna of New Zealand Ko te Aitanga Pepeke o Aotearoa Number / Nama 38 Naturalised terrestrial Stylommatophora (Mousca Gasooa Gay M ake acae eseac iae ag 317 amio ew eaa 4 Maaaki Whenua Ρ Ε S S ico Caeuy ew eaa 1999 Coyig © acae eseac ew eaa 1999 o a o is wok coee y coyig may e eouce o coie i ay om o y ay meas (gaic eecoic o mecaica icuig oocoyig ecoig aig iomaio eiea sysems o oewise wiou e wie emissio o e uise Caaoguig i uicaio AKE G Μ (Gay Micae 195— auase eesia Syommaooa (Mousca Gasooa / G Μ ake — ico Caeuy Maaaki Weua ess 1999 (aua o ew eaa ISS 111-533 ; o 3 IS -7-93-5 I ie 11 Seies UC 593(931 eae o uIicaio y e seies eio (a comee y eo Cosy usig comue-ase e ocessig ayou scaig a iig a acae eseac M Ae eseac Cee iae ag 917 Aucka ew eaa Māoi summay e y aco uaau Cosuas Weigo uise y Maaaki Weua ess acae eseac O o ico Caeuy Wesie //wwwmwessco/ ie y G i Weigo o coe eoceas eicuaum (ue a eigo oaa (owe (IIusao G M ake oucio o e coou Iaes was ue y e ew eaIa oey oa ue oeies eseac -

ABSTRACT Title of Dissertation: PATTERNS IN
ABSTRACT Title of Dissertation: PATTERNS IN DIVERSITY AND DISTRIBUTION OF BENTHIC MOLLUSCS ALONG A DEPTH GRADIENT IN THE BAHAMAS Michael Joseph Dowgiallo, Doctor of Philosophy, 2004 Dissertation directed by: Professor Marjorie L. Reaka-Kudla Department of Biology, UMCP Species richness and abundance of benthic bivalve and gastropod molluscs was determined over a depth gradient of 5 - 244 m at Lee Stocking Island, Bahamas by deploying replicate benthic collectors at five sites at 5 m, 14 m, 46 m, 153 m, and 244 m for six months beginning in December 1993. A total of 773 individual molluscs comprising at least 72 taxa were retrieved from the collectors. Analysis of the molluscan fauna that colonized the collectors showed overwhelmingly higher abundance and diversity at the 5 m, 14 m, and 46 m sites as compared to the deeper sites at 153 m and 244 m. Irradiance, temperature, and habitat heterogeneity all declined with depth, coincident with declines in the abundance and diversity of the molluscs. Herbivorous modes of feeding predominated (52%) and carnivorous modes of feeding were common (44%) over the range of depths studied at Lee Stocking Island, but mode of feeding did not change significantly over depth. One bivalve and one gastropod species showed a significant decline in body size with increasing depth. Analysis of data for 960 species of gastropod molluscs from the Western Atlantic Gastropod Database of the Academy of Natural Sciences (ANS) that have ranges including the Bahamas showed a positive correlation between body size of species of gastropods and their geographic ranges. There was also a positive correlation between depth range and the size of the geographic range. -

January 15, 2015
January 15, 2015 Below are: (1) a bibliography of works on western Atlantic marine mollusks appearing in the journal Avicennia . It includes a listing of all species-level taxa introduced in the cited paper. (2) An alphabetical list of taxa described (new 168, old 1), by family, in the cited papers. These databases are adapted from Gary Rosenberg's Malacolog 4.1.1 < http://www.malacolog.org/ > , and the latter was generated with major assistance from Peggy Williams of Tallevast, FL. Publication date refinement, orthographic emendations, synonymies, and generic reassignments are the work of Dr. Rosenberg. The purpose of this webfeature is to provide a searchable, Internet-linked resource now that the entirety of this discontinued journal (1993-2007) is available on-line at: http://www.biodiversitylibrary.org/bibliography/79640#/summary ************************************************************************************* Ardila, N. E. and P. Rachello. 2004. Opisthobranchs (Mollusca: Gastropoda) collected by the cruises Invemar-Macrofauna II in the Colombian Caribbean (20-150m). Avicennia 17: 57-66. [True date: pre 27 July.] [No species-group names included in Malacolog were introduced in this work.] Caballer, M. and J. Ortea. 2007. Nueva especie del género Hermaea Lovén, 1844 (Mollusca: Sacoglossa), de la costa norte de La Habana, Cuba. Avicennia 19 : 127-132. [Stated date: -- Sep 2007.] Hermaea nautica Herma Caballer, M., J. Ortea and J. Espinosa. 2001. Descripción de una nueva especie de Eubranchus Forbes, 1834. Avicennia, Suplemento 4 : 55-56, pl. 2. [True date: pre Nov 8.] Eubranchus leopoldoi Caballer, M., J. Ortea and J. Espinosa. 2006. Descripción de una nueva especie de Alderiopsis Baba, 1968. Avicennia 18 : 57-60. -

Recent Advances and Unanswered Questions in Deep Molluscan Phylogenetics Author(S): Kevin M
Recent Advances and Unanswered Questions in Deep Molluscan Phylogenetics Author(s): Kevin M. Kocot Source: American Malacological Bulletin, 31(1):195-208. 2013. Published By: American Malacological Society DOI: http://dx.doi.org/10.4003/006.031.0112 URL: http://www.bioone.org/doi/full/10.4003/006.031.0112 BioOne (www.bioone.org) is a nonprofit, online aggregation of core research in the biological, ecological, and environmental sciences. BioOne provides a sustainable online platform for over 170 journals and books published by nonprofit societies, associations, museums, institutions, and presses. Your use of this PDF, the BioOne Web site, and all posted and associated content indicates your acceptance of BioOne’s Terms of Use, available at www.bioone.org/page/terms_of_use. Usage of BioOne content is strictly limited to personal, educational, and non-commercial use. Commercial inquiries or rights and permissions requests should be directed to the individual publisher as copyright holder. BioOne sees sustainable scholarly publishing as an inherently collaborative enterprise connecting authors, nonprofit publishers, academic institutions, research libraries, and research funders in the common goal of maximizing access to critical research. Amer. Malac. Bull. 31(1): 195–208 (2013) Recent advances and unanswered questions in deep molluscan phylogenetics* Kevin M. Kocot Auburn University, Department of Biological Sciences, 101 Rouse Life Sciences, Auburn University, Auburn, Alabama 36849, U.S.A. Correspondence, Kevin M. Kocot: [email protected] Abstract. Despite the diversity and importance of Mollusca, evolutionary relationships among the eight major lineages have been a longstanding unanswered question in Malacology. Early molecular studies of deep molluscan phylogeny, largely based on nuclear ribosomal gene data, as well as morphological cladistic analyses largely failed to provide robust hypotheses of relationships among major lineages. -

Describing Species
DESCRIBING SPECIES Practical Taxonomic Procedure for Biologists Judith E. Winston COLUMBIA UNIVERSITY PRESS NEW YORK Columbia University Press Publishers Since 1893 New York Chichester, West Sussex Copyright © 1999 Columbia University Press All rights reserved Library of Congress Cataloging-in-Publication Data © Winston, Judith E. Describing species : practical taxonomic procedure for biologists / Judith E. Winston, p. cm. Includes bibliographical references and index. ISBN 0-231-06824-7 (alk. paper)—0-231-06825-5 (pbk.: alk. paper) 1. Biology—Classification. 2. Species. I. Title. QH83.W57 1999 570'.1'2—dc21 99-14019 Casebound editions of Columbia University Press books are printed on permanent and durable acid-free paper. Printed in the United States of America c 10 98765432 p 10 98765432 The Far Side by Gary Larson "I'm one of those species they describe as 'awkward on land." Gary Larson cartoon celebrates species description, an important and still unfinished aspect of taxonomy. THE FAR SIDE © 1988 FARWORKS, INC. Used by permission. All rights reserved. Universal Press Syndicate DESCRIBING SPECIES For my daughter, Eliza, who has grown up (andput up) with this book Contents List of Illustrations xiii List of Tables xvii Preface xix Part One: Introduction 1 CHAPTER 1. INTRODUCTION 3 Describing the Living World 3 Why Is Species Description Necessary? 4 How New Species Are Described 8 Scope and Organization of This Book 12 The Pleasures of Systematics 14 Sources CHAPTER 2. BIOLOGICAL NOMENCLATURE 19 Humans as Taxonomists 19 Biological Nomenclature 21 Folk Taxonomy 23 Binomial Nomenclature 25 Development of Codes of Nomenclature 26 The Current Codes of Nomenclature 50 Future of the Codes 36 Sources 39 Part Two: Recognizing Species 41 CHAPTER 3. -

Documents Félix A
Click Here & Upgrade Expanded Features PDF Unlimited Pages CompleteDocuments Félix A. Grana Raffucci. Junio, 2007. NOMENCLATURA DE LOS ORGANISMOS ACUÁTICOS Y MARINOS DE PUERTO RICO E ISLAS VÍRGENES. Volumen 4: Moluscos de Puerto Rico e Islas Vírgenes. Parte 3. Clase Gastropoda Órden Caenogastropoda Familias Eulimidae a Conidae Click Here & Upgrade Expanded Features PDF Unlimited Pages CompleteDocuments CLAVE DE COMENTARIOS: M= organismo reportado de ambientes marinos E= organismo reportado de ambientes estuarinos D= organismo reportado de ambientes dulceacuícolas int= organismo reportado de ambientes intermareales T= organismo reportado de ambientes terrestres L= organismo pelágico B= organismo bentónico P= organismo parasítico en alguna etapa de su vida F= organismo de valor pesquero Q= organismo de interés para el acuarismo A= organismo de interés para artesanías u orfebrería I= especie exótica introducida p=organismo reportado específicamente en Puerto Rico u= organismo reportado específicamente en las Islas Vírgenes de Estados Unidos b= organismo reportado específicamente en las Islas Vírgenes Británicas números= profundidades, en metros, en las que se ha reportado la especie Click Here & Upgrade Expanded Features PDF Unlimited Pages CompleteDocuments INDICE DE FAMILIAS EN ESTE VOLUMEN Aclididae Aclis Buccinidae Antillophos Bailya Belomitra Colubraria Engina Engoniophos Manaria Monostiolum Muricantharus Parviphos Pisania Pollia Cerithiopsidae Cerithiopsis Horologica Retilaskeya Seila Cancellariidae Agatrix Cancellaria Trigonostoma -
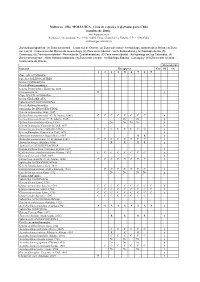
Moluscos - Filo MOLLUSCA
Moluscos - Filo MOLLUSCA. Lista de especies registradas para Cuba (octubre de 2006). José Espinosa Sáez Instituto de Oceanología, Ave 1ª No. 18406, Playa, Ciudad de La Habana, C.P. 11200, Cuba [email protected] Zonas biogeográficas: (1) Zona suroriental – Costa sur de Oriente, (2) Zona surcentral - Archipiélago Jardines de la Reina, (3) Zona sur central - Costa al sur del Macizo de Guamuhaya, (4) Zona suroccidental - Golfo de Batabanó y Archipiélago de los, (5) Canarreos, (6) Zona suroccidental - Península de Guanahacabibes, (7) Zona noroccidental - Archipiélago de Los Colorados, (8) Zona noroccidental - Norte Habana-Matanzas, (9) Zona norte-central - Archipiélago Sabana - Camagüey, (10) Zona norte-oriental - Costa norte de Oriente Abreviaturas Especies Bioegiones Cu Pl Oc 1 2 3 4 5 6 7 8 9 Clase APLACOPHORA Subclase SOLENOGASTRES Orden CAVIBELONIA Familia Proneomeniidae Género Proneomenia Hubrecht, 1880 Proneomenia sp . R x Clase POLYPLACOPHORA Orden NEOLORICATA Suborden ISCHNOCHITONINA Familia Ischnochitonidae Subfamilia ISCHNOCHITONINAE Género Ischnochiton Gray, 1847 Ischnochiton erythronotus (C. B. Adams, 1845) C C C C C C C C x Ischnochiton papillosus (C. B. Adams, 1845) Nc Nc x Ischnochiton striolatus (Gray, 1828) Nc Nc Nc Nc x Género Ischnoplax Carpenter in Dall, 1879 x Ischnoplax pectinatus (Sowerby, 1832) C C C C C C C C x Género Stenoplax Carpenter in Dall, 1879 x Stenoplax bahamensis Kaas y Belle, 1987 R R x Stenoplax purpurascens (C. B. Adams, 1845) C C C C C C C C x Stenoplax boogii (Haddon, 1886) R R R R x Subfamilia CALLISTOPLACINAE Género Callistochiton Carpenter in Dall, 1879 x Callistochiton shuttleworthianus Pilsbry, 1893 C C C C C C C C x Género Ceratozona Dall, 1882 x Ceratozona squalida (C. -

A Defense of a Sentiocentric Approach to Environmental Ethics
University of Tennessee, Knoxville TRACE: Tennessee Research and Creative Exchange Doctoral Dissertations Graduate School 8-2012 Minding Nature: A Defense of a Sentiocentric Approach to Environmental Ethics Joel P. MacClellan University of Tennessee, Knoxville, [email protected] Follow this and additional works at: https://trace.tennessee.edu/utk_graddiss Part of the Ethics and Political Philosophy Commons Recommended Citation MacClellan, Joel P., "Minding Nature: A Defense of a Sentiocentric Approach to Environmental Ethics. " PhD diss., University of Tennessee, 2012. https://trace.tennessee.edu/utk_graddiss/1433 This Dissertation is brought to you for free and open access by the Graduate School at TRACE: Tennessee Research and Creative Exchange. It has been accepted for inclusion in Doctoral Dissertations by an authorized administrator of TRACE: Tennessee Research and Creative Exchange. For more information, please contact [email protected]. To the Graduate Council: I am submitting herewith a dissertation written by Joel P. MacClellan entitled "Minding Nature: A Defense of a Sentiocentric Approach to Environmental Ethics." I have examined the final electronic copy of this dissertation for form and content and recommend that it be accepted in partial fulfillment of the equirr ements for the degree of Doctor of Philosophy, with a major in Philosophy. John Nolt, Major Professor We have read this dissertation and recommend its acceptance: Jon Garthoff, David Reidy, Dan Simberloff Accepted for the Council: Carolyn R. Hodges Vice Provost and Dean of the Graduate School (Original signatures are on file with official studentecor r ds.) MINDING NATURE: A DEFENSE OF A SENTIOCENTRIC APPROACH TO ENVIRONMENTAL ETHICS A Dissertation Presented for the Doctor of Philosophy Degree The University of Tennessee, Knoxville Joel Patrick MacClellan August 2012 ii The sedge is wither’d from the lake, And no birds sing. -

Shell Show 2010
! "#$%!&'!!!!!!!!!!(#%!)!!!!!!!!! #*+#,-.!)'/'! ! !"##$%&#" Page 3 EDITORIAL I have decided to keep this Editorial very brief and thank all the members who have contributed to this edition for their efforts in producing some very interesting reading. I have very much enjoyed the article by S.P. Dance on a “Necklace cone for Charles Kingsley” as I have in my own collection of cowry shells some specimens with very clear numbers appearing within the pattern. In the next issue I will take some photographs of these cowries to share with our members. Articles are always welcome for Pallidula; now that we have a full colour magazine, please do include photographs or other pictures with your articles – this makes for much more interesting reading. Please read carefully the Secretary’s Notes on the following page which includes some very important information about the upcoming events and changes within the Committee roles. Please keep up the good work with your articles and look forward to seeing you all at Theydon Bois for the October Shell Show. The Editor Dates for your Diary Plans are already in hand for future Club Meetings. Members may like to note the following dates:- Saturday 30th October 2010 - Shell Show at Theydon Bois Community Centre Saturday 30th April 2011 - Shell Convention at Theydon Bois Community Centre Saturday 17th September 2011 - Chatsworth House, Derbyshire Saturday 29th October 2011 - Shell Convention at Theydon Bois Community Centre Other event dates for 2011 will be announced in the April edition of Pallidula Please don’t forget to log onto THE BRITISH SHELL COLLECTORS CLUB website and check out our regular updates and articles www.britishshellclub.org.uk Conchological Society Indoor meetings are held in the Dorothea Bate Room (Palaeontology Demonstration Room) in the Natural History Museum, South Kensington, London, and consist of talks on a wide variety of conchological subjects. -

BASTERIA, 123-143, Oxychilus
BASTERIA, Vol. 61: 123-143, 1998 Oxychilus mortilleti (Pfeiffer, 1859): a redescription (Pulmonata, Zonitidae) G. Manganelli & F. Giusti Dipartimento di Biologia Evolutiva dell'Universita di Siena, Via P.A. Mattioli 4, 1-53100 Siena, Italy The taxonomic and nomenclatural of status Oxychilus mortilleti (Pfeiffer, 1859) is revised. O. mortilleti is a medium-sized Oxychilus species which canonly be distinguished from similar shelled, sympatric species [O. cellarius(Müller, 1774), O. draparnaudi(Beck, 1837), and O. adamii (Westerlund, 1886)], on the basis of the following anatomical characters: penis divided into proximal and distal constriction about three distal parts by (1), proximal penis times as long as penis (2), terminal, constricted part (‘bottle-neck’) of proximal penis long, slender, straight (occasionally slightly bent) (3), penis sheath covering almost entire distal penis (4). At present, O. mortilleti is only known from southern central Europe (Austria, Czech Republic and the Alps). In the Alps, confirmed records exist only for Ticino (Switzerland), Piedmont, Lombardy and Venetum (Italy). Key words: Gastropoda, Pulmonata, Zonitidae, Oxychilus mortilleti, redescription, taxonomy, nomenclature, Italy. INTRODUCTION Ten species of Oxychilus are reported from the Alps: O. adamii (Westerlund, 1886), O. alliarius (Miller, 1822), Oxychilus cellarius (Miiller, 1774), O. clarus (Held, 1838), O. depressus (Sterki, 1880), O. draparnaudi (Beck, 1837), O. glaber (Rossmassler, 1835), O. helveticus (Blum, 1881), O. mortilleti (Pfeiffer, 1859), and O. polygyra (Pollonera, 1885) (Riedel, 1980; Kerney et al., 1979, 1983; Falkner, 1990; Manganelli et al., 1995). Some of these species are well known because they have been redescribed recently: O. cellarius and O. draparnaudi by Giusti & Manganelli (1997), O. clarus by Giusti et al. -

Developing Perspectives on Molluscan Shells, Part 1: Introduction and Molecular Biology
CHAPTER 1 DEVELOPING PERSPECTIVES ON MOLLUSCAN SHELLS, PART 1: INTRODUCTION AND MOLECULAR BIOLOGY KEVIN M. KOCOT1, CARMEL MCDOUGALL, and BERNARD M. DEGNAN 1Present Address: Department of Biological Sciences and Alabama Museum of Natural History, The University of Alabama, Tuscaloosa, AL 35487, USA; E-mail: [email protected] School of Biological Sciences, The University of Queensland, St. Lucia, Queensland 4072, Australia CONTENTS Abstract ........................................................................................................2 1.1 Introduction .........................................................................................2 1.2 Insights From Genomics, Transcriptomics, and Proteomics ............13 1.3 Novelty in Molluscan Biomineralization ..........................................21 1.4 Conclusions and Open Questions .....................................................24 Keywords ...................................................................................................27 References ..................................................................................................27 2 Physiology of Molluscs Volume 1: A Collection of Selected Reviews ABSTRACT Molluscs (snails, slugs, clams, squid, chitons, etc.) are renowned for their highly complex and robust shells. Shell formation involves the controlled deposition of calcium carbonate within a framework of macromolecules that are secreted by the outer epithelium of a specialized organ called the mantle. Molluscan shells display remarkable morphological