WES Gene Package Disorders of Sex Development (DSD)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Bhagwan Moorjani, MD, FAAP, FAAN • Requires Knowledge of Normal CNS Developmental (I.E
1/16/2012 Neuroimaging in Childhood • Neuroimaging issues are distinct from Pediatric Neuroimaging in adults Neurometabolic-degenerative disorder • Sedation/anesthesia and Epilepsy • Motion artifacts Bhagwan Moorjani, MD, FAAP, FAAN • Requires knowledge of normal CNS developmental (i.e. myelin maturation) • Contrast media • Parental anxiety Diagnostic Approach Neuroimaging in Epilepsy • Age of onset • Peak incidence in childhood • Static vs Progressive • Occurs as a co-morbid condition in many – Look for treatable causes pediatric disorders (birth injury, – Do not overlook abuse, Manchausen if all is negative dysmorphism, chromosomal anomalies, • Phenotype presence (syndromic, HC, NCS, developmental delays/regression) systemic involvement) • Predominant symptom (epilepsy, DD, • Many neurologic disorders in children weakness/motor, psychomotor regression, have the same chief complaint cognitive/dementia) 1 1/16/2012 Congenital Malformation • Characterized by their anatomic features • Broad categories: based on embryogenesis – Stage 1: Dorsal Induction: Formation and closure of the neural tube. (Weeks 3-4) – Stage 2: Ventral Induction: Formation of the brain segments and face. (Weeks 5-10) – Stage 3: Migration and Histogenesis: (Months 2-5) – Stage 4: Myelination: (5-15 months; matures by 3 years) Dandy Walker Malformation Dandy walker • Criteria: – high position of tentorium – dysgenesis/agenesis of vermis – cystic dilatation of fourth ventricle • commonly associated features: – hypoplasia of cerebellum – scalloping of inner table of occipital bone • associated abnormalities: – hydrocephalus 75% – dysgenesis of corpus callosum 25% – heterotropia 10% 2 1/16/2012 Etiology of Epilepsy: Developmental and Genetic Classification of Gray Matter Heterotropia Cortical Dysplasia 1. Secondary to abnormal neuronal and • displaced masses of nerve cells • Subependymal glial proliferation/apoptosis (gray matter) heterotropia (most • most common: small nest common) 2. -

Lissencephaly
LE JOURNAL CANADIEN DES SCIENCES NEUROLOGIQUES Lissencephaly MARGARET G. NORMAN, MAUREEN ROBERTS, J. SIROIS, L. J. M. TREMBLAY SUMMARY: The first reported case of tic heterogeneity. Lissencephaly and INTRODUCTION lissencephaly resulting from a consan- pachygyria may eventually be shown to Lissencephaly (agyria) is charac guinous union strengthens the supposi be due to different causes, some inher terized by a smooth brain, without tion that in some cases, it is transmitted ited, some acquired. The classical ex sulci or gyri. The microscopic as an autosomal recessive trait. Com amples of lissencephaly are different parison of this case with a sporadically morphologically from a case in which an anatomy of the cortex varies, some occuring case of lissencephaly, with dif tenatal cytomegalovirus infection had cases showing no laminae, others ferent cortical morphology, suggests produced a small smooth brain. This four laminae. Associated abnormal - that lissencephaly may be an example of suggests that antenatal viral infections ities are masses of heterotopic grey either varying gene expressivity or gene- are destructive rather than teratogenic. matter around the ventricles. Heterotopias of the inferior olivary nuclei and cerebellar roof nuclei are RESUME: Le premier cas de ment, il sera peut-etre demontre que la frequently present. Pachygyria is a lissencephalie resultant d'une union lissencephalie et la pachygyrie sont dues morphologically similar anomaly in consanguine renforcit I'hypothese qu'au a des causes differentes, certaines etant which the brain has wide simple moins dans certain cas la lissencephalie hereditaires, d'autres etant acquises. est transmise par un trait autosomal Les examples classiques de gyri. Some have regarded lissence recessif. -

Supratentorial Brain Malformations
Supratentorial Brain Malformations Edward Yang, MD PhD Department of Radiology Boston Children’s Hospital 1 May 2015/ SPR 2015 Disclosures: Consultant, Corticometrics LLC Objectives 1) Review major steps in the morphogenesis of the supratentorial brain. 2) Categorize patterns of malformation that result from failure in these steps. 3) Discuss particular imaging features that assist in recognition of these malformations. 4) Reference some of the genetic bases for these malformations to be discussed in greater detail later in the session. Overview I. Schematic overview of brain development II. Abnormalities of hemispheric cleavage III. Commissural (Callosal) abnormalities IV. Migrational abnormalities - Gray matter heterotopia - Pachygyria/Lissencephaly - Focal cortical dysplasia - Transpial migration - Polymicrogyria V. Global abnormalities in size (proliferation) VI. Fetal Life and Myelination Considerations I. Schematic Overview of Brain Development Embryology Top Mid-sagittal Top Mid-sagittal Closed Neural Tube (4 weeks) Corpus Callosum Callosum Formation Genu ! Splenium Cerebral Hemisphere (11-20 weeks) Hemispheric Cleavage (4-6 weeks) Neuronal Migration Ventricular/Subventricular Zones Ventricle ! Cortex (8-24 weeks) Neuronal Precursor Generation (Proliferation) (6-16 weeks) Embryology From ten Donkelaar Clinical Neuroembryology 2010 4mo 6mo 8mo term II. Abnormalities of Hemispheric Cleavage Holoprosencephaly (HPE) Top Mid-sagittal Imaging features: Incomplete hemispheric separation + 1)1) No septum pellucidum in any HPEs Closed Neural -

Spectrum of Clinical and Associated MR Imaging Findings in Children with Olfactory Anomalies
Published March 17, 2016 as 10.3174/ajnr.A4738 ORIGINAL RESEARCH PEDIATRICS Spectrum of Clinical and Associated MR Imaging Findings in Children with Olfactory Anomalies X T.N. Booth and X N.K. Rollins ABSTRACT BACKGROUND AND PURPOSE: The olfactory apparatus, consisting of the bulb and tract, is readily identifiable on MR imaging. Anom- alous development of the olfactory apparatus may be the harbinger of anomalies of the secondary olfactory cortex and associated structures. We report a large single-site series of associated MR imaging findings in patients with olfactory anomalies. MATERIALS AND METHODS: A retrospective search of radiologic reports (2010 through 2014) was performed by using the keyword “olfactory”; MR imaging studies were reviewed for olfactory anomalies and intracranial and skull base malformations. Medical records were reviewed for clinical symptoms, neuroendocrine dysfunction, syndromic associations, and genetics. RESULTS: We identified 41 patients with olfactory anomalies (range, 0.03–18 years of age; M/F ratio, 19:22); olfactory anomalies were bilateral in 31 of 41 patients (76%) and absent olfactory bulbs and olfactory tracts were found in 56 of 82 (68%). Developmental delay was found in 24 (59%), and seizures, in 14 (34%). Pituitary dysfunction was present in 14 (34%), 8 had panhypopituitarism, and 2 had isolated hypogonadotropic hypogonadism. CNS anomalies, seen in 95% of patients, included hippocampal dysplasia in 26, cortical malformations in 15, malformed corpus callosum in 10, and optic pathway hypoplasia in 12. Infratentorial anomalies were seen in 15 (37%) patients and included an abnormal brain stem in 9 and an abnormal cerebellum in 3. Four patients had an abnormal membranous labyrinth. -

Blueprint Genetics Neuronal Migration Disorder Panel
Neuronal Migration Disorder Panel Test code: MA2601 Is a 59 gene panel that includes assessment of non-coding variants. Is ideal for patients with a clinical suspicion of neuronal migration disorder. About Neuronal Migration Disorder Neuronal migration disorders (NMDs) are a group of birth defects caused by the abnormal migration of neurons in the developing brain and nervous system. During development, neurons must migrate from the areas where they are originate to the areas where they will settle into their proper neural circuits. The structural abnormalities found in NMDs include schizencephaly, porencephaly, lissencephaly, agyria, macrogyria, polymicrogyria, pachygyria, microgyria, micropolygyria, neuronal heterotopias, agenesis of the corpus callosum, and agenesis of the cranial nerves. Mutations of many genes are involved in neuronal migration disorders, such as DCX in classical lissencephaly spectrum, TUBA1A in microlissencephaly with agenesis of the corpus callosum, and RELN and VLDLR in lissencephaly with cerebellar hypoplasia. Mutations in ARX cause a variety of phenotypes ranging from hydranencephaly or lissencephaly to early-onset epileptic encephalopathies, including Ohtahara syndrome and infantile spasms or intellectual disability with no brain malformations. Availability 4 weeks Gene Set Description Genes in the Neuronal Migration Disorder Panel and their clinical significance Gene Associated phenotypes Inheritance ClinVar HGMD ACTB* Baraitser-Winter syndrome AD 55 60 ACTG1* Deafness, Baraitser-Winter syndrome AD 27 47 ADGRG1 -

Miller–Dieker Syndrome, Type 1 Lissencephaly
Journal of Perinatology (2008) 28, 313–315 r 2008 Nature Publishing Group All rights reserved. 0743-8346/08 $30 www.nature.com/jp IMAGING CASE BOOK Miller–Dieker syndrome, type 1 lissencephaly TE Herman and MJ Siegel Department of Radiology, Mallinckrodt Institute of Radiology, Washington University School of Medicine, St Louis, MO, USA Journal of Perinatology (2008) 28, 313–315; doi:10.1038/sj.jp.7211920 bitemporal shallowness. The ears were low set. Retromicrognathia, and microphthalmia, more marked on the left, were also present. Case presentation The child had a cranial sonogram (Figure 1) and cranial magnetic A 1910 g infant was born at 41-weeks gestation to a 19-year-old resonance imaging (Figure 2). gravida 1 mother after a pregnancy complicated by hydramnios and poor fetal movement. The child was delivered by a cesarean section because of fetal decelerations with labor. Severe intrauterine Denouement and discussion growth retardation was present with the infant’s weight, length and The cranial sonogram demonstrated an abnormally smooth brain head circumference all less than the tenth percentile for gestational with no gyration or sulcation with a wide, shallow sylvian fissure age. The patient’s head demonstrated a wasted appearance with creating a figure-of-8 appearance. The absence of all gyri is Figure 1 (a) Coronal cranial sonogram at the level of the third ventricle demonstrating a figure-of-8 appearance of the brain with wide sylvian fissure and with no sulci or gyri. (b) Sagittal right lateral cranial sonogram demonstrating absence of gyri over the ipsilateral hemisphere. (c) Right sagittal sonogram demonstrating echogenic linear thalamostriate vessels (arrow). -

Medical Diagnosis/Conditions for Eligibility in AEIS
Medical Diagnosis/Conditions for Eligibility in AEIS 1) Achondroplasia 2) Agenesis of Corpus Callosum 3) Agyria (Lissencephaly) 4) Albinism 5) Amniotic Band syndrome 6) Anencephaly 7) Angelman’s syndrome 8) Anophthalmia 9) Apert syndrome 10) Aplasia of the brain (brain malformation/abnormality) 11) Arhinencephaly (Holoprosencephaly) 12) Arnold-Chiari syndrome 13) Arthrogryposis 14) Asperger syndrome/disorder 15) Asphyxiating Thoracic Dystrophy (Jeune syndrome) 16) Attachment disorder 17) Autism/Autism Spectrum disorder 18) Bardet-Biedl syndrome 19) Brain injury/degeneration 20) Brain malformation/abnormality 21) Cerebral Palsy (all types) 22) CHARGE syndrome 23) Chiari Malformation 24) Childhood Depression 25) Childhood Disintegrative disorder 26) Cornelia de Lange syndrome 27) Cortical vision impairment (vision loss/impairment) 28) Cri-du-Chat syndrome 29) Cytomegalovirus (CMV) 30) Dandy Walker syndrome/variant 31) De Morsier syndrome (Septo-Optic Dysplasia) 32) Developmental Apraxia 33) DiGeorge syndrome 34) Dilantin syndrome (Fetal Hydantoin syndrome) 35) Down Syndrome (Trisomy 21) 36) Edwards syndrome (Trisomy 18) 37) Encephalomalacia 38) Encephalopathy 39) Epilepsy (seizure disorder) 40) Fetal Alcohol syndrome 41) Fetal Hydantoin syndrome (Dilantin syndrome) 42) Fragile X syndrome 43) Genetic/Chromosomal malformation/abnormality (not listed) 44) Hearing Loss/Impairment 45) Heart Disease/Defect (not listed) 46) Hemiplegia 47) Herpes Simplex Virus (HSV) 48) Holoprosencephaly (Arhinencephaly) 49) Holt Oram syndrome 50) Hydraencephaly -

Disorders of Sex Development Panel
Abnormal Genitalia/ Disorders of Sex Development Panel Test code: EN0201 Is a 62 gene panel that includes assessment of non-coding variants. Is ideal for patients presenting with ambiguous genitalia, patients suspected to have a disorder of sexual development and patients suspected to have congenital adrenal hyperplasia (CAH). About Abnormal Genitalia/ Disorders of Sex Development Disorders of sex development (DSD) are a group of congenital conditions characterized by problems in the course of gender patterning, gonadal and sex development. It has been estimated that 1% – 2% of live births have some aspect of DSD. Approximately 5% of infants with DSD have ambiguous genitalia and indeterminate sex at birth. However, the vast majority of these patients do not require corrective surgery. Patients with 46,XY DSD have often impaired androgen synthesis or action and may have normal female external genitalia, while patients with 46,XX DSD conditions have often androgen excess. In 46,XX females, congenital adrenal hyperplasia (CAH) caused by 21-hydroxylase deficiency (21-OHD) is the most common cause of DSD. The estimated prevalence of CAH is 1:10,000 and 90%-95% of cases are due to mutations in CYP21A2. The severity of the condition often depends on the residual enzyme activity subdiving CYP21A2 mutations in severe (classic phenotype, enzyme activity 0%-10%) and mild (non-classic, enzyme activity 20%-50%). Androgen insensitivity syndrome (AIS), caused by mutations in AR, is characterized by feminization of external genitalia and atypical sexual development in 46,XY individuals. The condition may be complete, partial or mild, depending on the level of androgen insensitivity. -

Syndromes with Lissencephaly
JMed Genet 1996;33:319-323 319 Syndrome of the month J Med Genet: first published as 10.1136/jmg.33.4.319 on 1 April 1996. Downloaded from Syndromes with lissencephaly D T Pilz, 0 W J Quarrell Earl Walker's paper in 1942 represents a de- logical types, assigned these to previously de- tailed review of early described cases of lis- scribed cases or syndromes, and discussed sencephaly and states that Owen (On the possible genetic mechanisms.67 The two main Institute of Medical anatomy of vertebrates, vol 3, London: Genetics, Long- pathological types he described provide a useful University Hospital of mans, Green & Co, 1868) is said to have basis on which to review "syndromes with lis- Wales, introduced the term lissencephaly to describe sencephaly". Heath Park, an agyric brain, from the Greek words "lissos" Cardiff CF4 4XN, UK D T Pilz (smooth) and "encephalus" (brain).' Key word: lissencephaly. Further reports of lissencephaly followed by Centre for Human Miller,2 Dieker et al,3 Warburg,45 and others and Genetics, their contributions are recognised in syndromes Type I or classical lissencephaly 117 Manchester Road, PREVALENCE Sheffield S10 5DN, UK now known as Miller-Dieker syndrome and 0 W J Quarrell Walker-Warburg syndrome. The only epidemiological data on the pre- valence of type I lissencephaly come from The Correspondence to: In his detailed analysis in 1984/85, Dobyns Dr Pilz. categorised lissencephaly into different patho- Netherlands, with 11-7 per million births.8 PATHOLOGY AND NEUROIMAGING Type I lissencephaly results from a neuro- migrational arrest between 12 and 16 weeks' gestation, and histologically the cortex has four instead ofsix layers. -

EUROCAT Syndrome Guide
JRC - Central Registry european surveillance of congenital anomalies EUROCAT Syndrome Guide Definition and Coding of Syndromes Version July 2017 Revised in 2016 by Ingeborg Barisic, approved by the Coding & Classification Committee in 2017: Ester Garne, Diana Wellesley, David Tucker, Jorieke Bergman and Ingeborg Barisic Revised 2008 by Ingeborg Barisic, Helen Dolk and Ester Garne and discussed and approved by the Coding & Classification Committee 2008: Elisa Calzolari, Diana Wellesley, David Tucker, Ingeborg Barisic, Ester Garne The list of syndromes contained in the previous EUROCAT “Guide to the Coding of Eponyms and Syndromes” (Josephine Weatherall, 1979) was revised by Ingeborg Barisic, Helen Dolk, Ester Garne, Claude Stoll and Diana Wellesley at a meeting in London in November 2003. Approved by the members EUROCAT Coding & Classification Committee 2004: Ingeborg Barisic, Elisa Calzolari, Ester Garne, Annukka Ritvanen, Claude Stoll, Diana Wellesley 1 TABLE OF CONTENTS Introduction and Definitions 6 Coding Notes and Explanation of Guide 10 List of conditions to be coded in the syndrome field 13 List of conditions which should not be coded as syndromes 14 Syndromes – monogenic or unknown etiology Aarskog syndrome 18 Acrocephalopolysyndactyly (all types) 19 Alagille syndrome 20 Alport syndrome 21 Angelman syndrome 22 Aniridia-Wilms tumor syndrome, WAGR 23 Apert syndrome 24 Bardet-Biedl syndrome 25 Beckwith-Wiedemann syndrome (EMG syndrome) 26 Blepharophimosis-ptosis syndrome 28 Branchiootorenal syndrome (Melnick-Fraser syndrome) 29 CHARGE -

Genetic Testing Medical Policy – Genetics
Genetic Testing Medical Policy – Genetics Please complete all appropriate questions fully. Suggested medical record documentation: • Current History & Physical • Progress Notes • Family Genetic History • Genetic Counseling Evaluation *Failure to include suggested medical record documentation may result in delay or possible denial of request. PATIENT INFORMATION Name: Member ID: Group ID: PROCEDURE INFORMATION Genetic Counseling performed: c Yes c No **Please check the requested analyte(s), identify number of units requested, and provide indication/rationale for testing. 81400 Molecular Pathology Level 1 Units _____ c ACADM (acyl-CoA dehydrogenase, C-4 to C-12 straight chain, MCAD) (e.g., medium chain acyl dehydrogenase deficiency), K304E variant _____ c ACE (angiotensin converting enzyme) (e.g., hereditary blood pressure regulation), insertion/deletion variant _____ c AGTR1 (angiotensin II receptor, type 1) (e.g., essential hypertension), 1166A>C variant _____ c BCKDHA (branched chain keto acid dehydrogenase E1, alpha polypeptide) (e.g., maple syrup urine disease, type 1A), Y438N variant _____ c CCR5 (chemokine C-C motif receptor 5) (e.g., HIV resistance), 32-bp deletion mutation/794 825del32 deletion _____ c CLRN1 (clarin 1) (e.g., Usher syndrome, type 3), N48K variant _____ c DPYD (dihydropyrimidine dehydrogenase) (e.g., 5-fluorouracil/5-FU and capecitabine drug metabolism), IVS14+1G>A variant _____ c F13B (coagulation factor XIII, B polypeptide) (e.g., hereditary hypercoagulability), V34L variant _____ c F2 (coagulation factor 2) (e.g., -
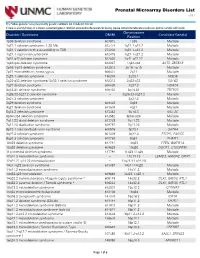
Prenatal Microarray Disorders List V19.1
Prenatal Microarray Disorders List v19.1 This "whole genome" array may identify genetic conditions not included in this list. If there is a family history of a known suspected genetic condition unrelated to the reason for testing, please contact the laboratory to discuss prior to sample submission. Chromosome Disorder / Syndrome OMIM Candidate Gene(s) Position 1p36 deletion syndrome 607872 1p36 Multiple 1q21.1 deletion syndrome, 1.35 Mb 612474 1q21.1-q21.2 Multiple 1q21.1 deletion with susceptibility to TAR 274000 1q21.1-q21.2 Multiple 1q21.1 duplication syndrome 612475 1q21.1-q21.2 Multiple 1q41-q42 deletion syndrome 612530 1q41-q42.12 Multiple 1q43-q44 deletion syndrome 612337 1q43-q44 AKT3, ZBTB18 2p16.1-p15 deletion syndrome 612513 2p16.1-p15 Multiple 2p21 microdeletion, homozygous 606407 2p21 Multiple 2q23.1 deletion syndrome 156200 2q23.1 MBD5 2q32-q33 deletion syndrome/ 2q33.1 deletion syndrome 612313 2q32-q33 SATB2 2q37 deletion syndrome 600430 2q37.3 HDAC4 3q13.31 deletion syndrome 615433 3q13.31 ZBTB20 3q26.33-3q27.2 deletion syndrome -- 3q26.33-3q27.2 Multiple 3q27.3 deletion syndrome -- 3q27.3 Multiple 3q29 deletion syndrome 609425 3q29 Multiple 4q21 deletion syndrome 613509 4q21 Multiple 5q14.3 deletion syndrome 613443 5q14.3 MEF2C 6pter-p24 deletion syndrome 612582 6pter-p24 Multiple 7q11.23 distal deletion syndrome 613729 7q11.23 Multiple 7q11.23 duplication syndrome 609757 7q11.23 Multiple 8p23.1 deletion/duplication syndrome 600576 8p23.1 GATA4 9q22.3 deletion syndrome 601309 9q22.3 PTCH1, FANCC 9q34.3 deletion syndrome