Full Prescribing Information [FDA]
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Targeting the Epidermal Growth Factor Receptor in EGFR-Mutated Lung Cancer: Current and Emerging Therapies
cancers Review Targeting the Epidermal Growth Factor Receptor in EGFR-Mutated Lung Cancer: Current and Emerging Therapies Karam Khaddour 1,*, Sushma Jonna 1, Alexander Deneka 2 , Jyoti D. Patel 3, Mohamed E. Abazeed 4, Erica Golemis 2 , Hossein Borghaei 5 and Yanis Boumber 3,6,* 1 Division of Hematology and Oncology, University of Illinois at Chicago, Chicago, IL 60612, USA; [email protected] 2 Fox Chase Cancer Center, Program in Molecular Therapeutics, Philadelphia, PA 19111, USA; [email protected] (A.D.); [email protected] (E.G.) 3 Robert H. Lurie Comprehensive Cancer Center, Division of Hematology/Oncology, Feinberg School of Medicine, Northwestern University, Chicago, IL 60611, USA; [email protected] 4 Robert H. Lurie Comprehensive Cancer Center, Department of Radiation Oncology, Feinberg School of Medicine, Northwestern University, Chicago, IL 60611, USA; [email protected] 5 Fox Chase Cancer Center, Department of Hematology and Oncology, Philadelphia, PA 19111, USA; [email protected] 6 Institute of Fundamental Medicine and Biology, Kazan Federal University, 420008 Kazan, Russia * Correspondence: [email protected] (K.K.); [email protected] (Y.B.) Simple Summary: Epidermal growth factor receptor (EGFR) mutations occur in a significant number Citation: Khaddour, K.; Jonna, S.; of lung cancer patients. Treatment outcomes in this subset of patients has greatly improved over the Deneka, A.; Patel, J.D.; Abazeed, M.E.; last decade after the introduction of EGFR tyrosine kinase inhibitors (TKIs), which demonstrated high Golemis, E.; Borghaei, H.; Boumber, Y. efficacy and improved survival in randomized clinical trials. Although EGFR TKIs became the stan- Targeting the Epidermal Growth dard of care in patients with EGFR-mutated lung cancer, resistance almost inevitably develops. -

Interim Report for the First Quarter of 2020
Genmab Announces Financial Results for the First Quarter of 2020 May 6, 2020; Copenhagen, Denmark; Interim Report for the First Quarter Ended March 31, 2020 Highlights DARZALEX® (daratumumab) net sales increased approximately 49% compared to the first quarter of 2019 to USD 937 million, resulting in royalty income of DKK 775 million DARZALEX approved in Europe in combination with bortezomib, thalidomide and dexamethasone for the treatment of adult patients with newly diagnosed multiple myeloma who are eligible for autologous stem cell transplant U.S. FDA approved TEPEZZA™ (teprotumumab-trbw), developed and commercialized by Horizon Therapeutics, for thyroid eye disease U.S. FDA accepted, with priority review, Novartis’ supplemental Biologics License Application for subcutaneous ofatumumab in relapsing multiple sclerosis Anthony Pagano appointed Chief Financial Officer Anthony Mancini appointed Chief Operating Officer “Despite the unprecedented challenges posed by the coronavirus (COVID-19) pandemic, we will continue to invest in our innovative proprietary products, technologies and capabilities and use our world-class expertise in antibody drug development to create truly differentiated products with the potential to help cancer patients. While Genmab is closely monitoring the developments in the rapidly evolving landscape, we are extremely fortunate to have a solid financial foundation and a fabulous and committed team to carry us through these uncertain times,” said Jan van de Winkel, Ph.D., Chief Executive Officer of Genmab. Financial Performance First Quarter of 2020 Revenue was DKK 892 million in the first quarter of 2020 compared to DKK 591 million in the first quarter of 2019. The increase of DKK 301 million, or 51%, was mainly driven by higher DARZALEX royalties. -
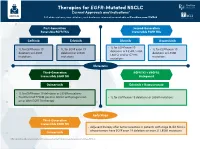
Therapies for EGFR-Mutated NSCLC Current Approvals and Indications1 Full Abbreviations, Accreditation, and Disclosure Information Available at Peerview.Com/CWE40
Therapies for EGFR-Mutated NSCLC 1 Current Approvals and Indications Full abbreviations, accreditation, and disclosure information available at PeerView.com/CWE40 First-Generation Second-Generation Reversible EGFR TKIs Irreversible EGFR TKIs Getinib Erlotinib Afatinib Dacomitinib • 1L for EGFR exon 19 • 1L for EGFR exon 19 • 1L for EGFR exon 19 • 1L for EGFR exon 19 deletions or L858R, S768I, deletions or L858R deletions or L858R deletions or L858R L861Q, and/or G719X mutations mutations mutations mutations Metastatic Third-Generation EGFR TKI + VEGFR2 Irreversible EGFR TKI Antagonist Osimertinib Erlotinib + Ramucirumab • 1L for EGFR exon 19 deletions or L858R mutations • Treatment of T790M-positive NSCLC with progression • 1L for EGFR exon 19 deletions or L858R mutations on or after EGFR TKI therapy Early Stage Third-Generation Irreversible EGFR TKI • Adjuvant therapy after tumor resection in patients with stage IB-IIIA NSCLC whose tumors have EGFR exon 19 deletions or exon 21 L858R mutations Osimertinib 1. https://www.fda.gov/drugs/resources-information-approved-drugs/hematologyoncology-cancer-approvals-safety-notifications. Molecular Testing Guidelines for NSCLC Latest Updates, Best Practices, and Patient-Reported Insights1 Full abbreviations, accreditation, and disclosure information available at PeerView.com/CWE40 Why Test Lung Cancer Patients for Genomic Alterations? • Genomic alterations are common in nonsquamous NSCLC (approximately 50%) • Targeted therapies produce better treatment outcomes (eg, higher response rates, improved -

Antibodies to Watch in 2021 Hélène Kaplona and Janice M
MABS 2021, VOL. 13, NO. 1, e1860476 (34 pages) https://doi.org/10.1080/19420862.2020.1860476 PERSPECTIVE Antibodies to watch in 2021 Hélène Kaplona and Janice M. Reichert b aInstitut De Recherches Internationales Servier, Translational Medicine Department, Suresnes, France; bThe Antibody Society, Inc., Framingham, MA, USA ABSTRACT ARTICLE HISTORY In this 12th annual installment of the Antibodies to Watch article series, we discuss key events in antibody Received 1 December 2020 therapeutics development that occurred in 2020 and forecast events that might occur in 2021. The Accepted 1 December 2020 coronavirus disease 2019 (COVID-19) pandemic posed an array of challenges and opportunities to the KEYWORDS healthcare system in 2020, and it will continue to do so in 2021. Remarkably, by late November 2020, two Antibody therapeutics; anti-SARS-CoV antibody products, bamlanivimab and the casirivimab and imdevimab cocktail, were cancer; COVID-19; Food and authorized for emergency use by the US Food and Drug Administration (FDA) and the repurposed Drug Administration; antibodies levilimab and itolizumab had been registered for emergency use as treatments for COVID-19 European Medicines Agency; in Russia and India, respectively. Despite the pandemic, 10 antibody therapeutics had been granted the immune-mediated disorders; first approval in the US or EU in 2020, as of November, and 2 more (tanezumab and margetuximab) may Sars-CoV-2 be granted approvals in December 2020.* In addition, prolgolimab and olokizumab had been granted first approvals in Russia and cetuximab saratolacan sodium was first approved in Japan. The number of approvals in 2021 may set a record, as marketing applications for 16 investigational antibody therapeutics are already undergoing regulatory review by either the FDA or the European Medicines Agency. -

In This Issue
IN THIS ISSUE PD-1 Blockade Is Not Unsafe in Patients with Lung Cancer and COVID-19 • COVID-19 severity and mortality were not • Although PD-1 blockade was not a • This suggests that PD-1 blockade increased in patients with lung cancer risk factor in this population, severity should be used when indicated in patients who received prior PD-1 blockade . and mortality in this group were high . with lung cancer despite COVID-19 . A key issue in oncology of whether patients received the immunotherapy recently or practice during the COVID-19 at any point prior to infection. There was a slight, statistically pandemic is whether PD-1 insignifi cant numeric increase in severity with PD-1 blockade, blockade affects the severity of but controlling for smoking history—which is an expected COVID-19 in patients with can- imbalance in receipt of prior PD-1 blockade and a risk factor cer. PD-1 blockade may increase for severe COVID-19 outcomes—abolished this correlation. COVID-19 severity by contrib- This work suggests that prior PD-1 blockade is not a clinically uting to hyperactive immune meaningful risk factor for worsened COVID-19 outcomes, res ponses to SARS-CoV-2 infec- implying that PD-1 blockade should be used when indicated tion—or, alternatively, may reduce despite the pandemic. Larger studies are needed to more fully severity by enhancing control of initial viral infection. To examine this question. Finally, it should be noted that, in minimize lung cancer as a confounder, Luo and colleagues this study population, more than half of patients with lung assessed the outcomes of concurrent COVID-19 and lung cancer and COVID-19 were hospitalized and almost half of cancer in 69 consecutive patients at a single institution in those hospitalized died, emphasizing the need for studies to New York City. -

Brief Report Preclinical Characterization of Mobocertinib
bioRxiv preprint doi: https://doi.org/10.1101/2020.09.28.317560; this version posted September 28, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Brief Report Preclinical characterization of mobocertinib highlights the putative therapeutic window of this novel EGFR inhibitor to EGFR exon 20 insertion mutations Pedro E. N. S. Vasconcelos1*; Ikei S. Kobayashi1*; Susumu S. Kobayashi1,2; Daniel B. Costa1* 1 Department of Medicine, Division of Hematology/Oncology, Harvard Medical School, Boston, MA, United States of America; 2 Division of Translational Genomics, Exploratory Oncology Research and Clinical Trial Center, National Cancer Center, Kashiwa, Chiba, Japan. * these authors contributed equally Correspondence to: *Daniel B. Costa, MD, PhD - Division of Medical Oncology, Beth Israel Deaconess Medical Center, 330 Brookline Av., Boston, MA 02215; Phone: 617-667-9236, Fax: 617-975-5665, Email: [email protected] Running Title: Mobocertinib for EGFR exon 20 mutations Category: Brief Report Word count: abstract (246), text (1989), references (15) Figures: 2 Tables: 1 Keywords: lung cancer; EGFR exon 20 insertion; mobocertinib; C797S; ERBB2 Acknowledgements/Funding: This work was funded in part through National Institutes of Health (NIH)/National Cancer Institute (NCI) grants R37 CA218707 (to D. B. Costa), R01 CA240257 (to S. S. Kobayashi) plus Department of Defense LC170223 (to S. S. Kobayashi) Disclosure of Potential Conflicts of interest: DBC reports personal fees (consulting fees and honoraria) and nonfinancial support (institutional research support) from Takeda/Millennium Pharmaceuticals, AstraZeneca, Pfizer and BluePrint Medicine, as well as nonfinancial support (institutional research support) from Merck Sharp and Dohme Corporation, Merrimack Pharmaceuticals, Bristol-Myers Squibb, Clovis Oncology, Spectrum Pharmaceuticals, Tesaro, all outside the submitted work. -

Antibodies to Watch in 2021 Hélène Kaplona and Janice M
MABS 2021, VOL. 13, NO. 1, e1860476 (34 pages) https://doi.org/10.1080/19420862.2020.1860476 PERSPECTIVE Antibodies to watch in 2021 Hélène Kaplona and Janice M. Reichert b aInstitut De Recherches Internationales Servier, Translational Medicine Department, Suresnes, France; bThe Antibody Society, Inc., Framingham, MA, USA ABSTRACT ARTICLE HISTORY In this 12th annual installment of the Antibodies to Watch article series, we discuss key events in antibody Received 1 December 2020 therapeutics development that occurred in 2020 and forecast events that might occur in 2021. The Accepted 1 December 2020 coronavirus disease 2019 (COVID-19) pandemic posed an array of challenges and opportunities to the KEYWORDS healthcare system in 2020, and it will continue to do so in 2021. Remarkably, by late November 2020, two Antibody therapeutics; anti-SARS-CoV antibody products, bamlanivimab and the casirivimab and imdevimab cocktail, were cancer; COVID-19; Food and authorized for emergency use by the US Food and Drug Administration (FDA) and the repurposed Drug Administration; antibodies levilimab and itolizumab had been registered for emergency use as treatments for COVID-19 European Medicines Agency; in Russia and India, respectively. Despite the pandemic, 10 antibody therapeutics had been granted the immune-mediated disorders; first approval in the US or EU in 2020, as of November, and 2 more (tanezumab and margetuximab) may Sars-CoV-2 be granted approvals in December 2020.* In addition, prolgolimab and olokizumab had been granted first approvals in Russia and cetuximab saratolacan sodium was first approved in Japan. The number of approvals in 2021 may set a record, as marketing applications for 16 investigational antibody therapeutics are already undergoing regulatory review by either the FDA or the European Medicines Agency. -

Amivantamab: a Potent Novel EGFR/C-MET Bispecific Antibody Therapy for EGFR-Mutated Non-Small Cell Lung Cancer
Review Lung Cancer Amivantamab: A Potent Novel EGFR/c-MET Bispecific Antibody Therapy for EGFR-mutated Non-small Cell Lung Cancer Matthew Z Guo,1 Kristen A Marrone,1 Alexander Spira,1,2 Kristine Freeman1,2 and Susan C Scott1 1. Johns Hopkins School of Medicine, Sidney Kimmel Comprehensive Cancer Center, Baltimore, MD, USA; 2. Virginia Cancer Specialists Research Institute, Fairfax, VA, USA; US Oncology Research, The Woodlands TX, USA hile the majority of epidermal growth factor receptor (EGFR) driver mutations in non-small cell lung cancer (NSCLC) are sensitive to treatment with targeted tyrosine kinase inhibitors (TKIs), anti-EGFR TKIs are susceptible to resistance mechanisms through Wsecondary mutations and also lack efficacy against tumours with non-classical EGFR mutations like exon 20 insertions. EGFR exon 20 insertions represent a challenging molecular subtype of NSCLC with no approved targeted therapy options and poor overall prognosis. Similarly, for NSCLC with EGFR mutations that are sensitive to TKIs, there are no approved targeted therapies following progression on a third-generation TKI without an alternative actionable mutation. Amivantamab is a novel, fully human bispecific anti-EGFR/c-MET antibody with clinical efficacy and favourable toxicity against both pre-treated EGFR exon 20 insertion NSCLC and classical EGFR mutations following development of TKI resistance. Preliminary findings from the phase I CHRYSALIS study report a 40% objective response rate with a median duration of response of 11.1 months for patients with pre-treated EGFR exon 20 insertion mutated tumours after platinum- based chemotherapy. Based on these results, the US Food and Drug Administration granted amivantamab accelerated approval in 2021 for treatment of adult patients with locally advanced or metastatic NSCLC with EGFR exon 20 insertion mutations whose disease has progressed on or after platinum-based chemotherapy. -

Treating Non-Small Cell Lung Cancer
cancer.org | 1.800.227.2345 Treating Non-Small Cell Lung Cancer If you've been diagnosed with non-small cell lung cancer (NSCLC), your cancer care team will discuss your treatment options with you. It's important to weigh the benefits of each treatment option against the possible risks and side effects. How is non-small cell lung cancer treated? Treatments for NSCLC can include: ● Surgery for Non-Small Cell Lung Cancer ● Radiofrequency Ablation (RFA) for Non-Small Cell Lung Cancer ● Radiation Therapy for Non-Small Cell Lung Cancer ● Chemotherapy for Non-Small Cell Lung Cancer ● Targeted Drug Therapy for Non-Small Cell Lung Cancer ● Immunotherapy for Non-Small Cell Lung Cancer ● Palliative Procedures for Non-Small Cell Lung Cancer Common treatment approaches The treatment options for non-small cell lung cancer (NSCLC) are based mainly on the stage (extent) of the cancer, but other factors, such as a person’s overall health and lung function, as well as certain traits of the cancer itself, are also important. In many cases, more than one of type of treatment is used. ● Treatment Choices for Non-Small Cell Lung Cancer, by Stage Who treats non-small cell lung cancer? You may have different types of doctors on your treatment team, depending on the 1 ____________________________________________________________________________________American Cancer Society cancer.org | 1.800.227.2345 stage of your cancer and your treatment options. These doctors could include: ● A thoracic surgeon: a doctor who treats diseases of the lungs and chest with surgery ● A radiation oncologist: a doctor who treats cancer with radiation therapy ● A medical oncologist: a doctor who treats cancer with medicines such as chemotherapy, targeted therapy, and immunotherapy ● A pulmonologist: a doctor who specializes in medical treatment of diseases of the lungs Many other specialists may be involved in your care as well, including nurse practitioners, nurses, psychologists, social workers, rehabilitation specialists, and other health professionals. -

Progress in EGFR-Mutant NSCLC: Where Are We Going? Transcript from a Touchexpert OPINIONS
touchEXPERT OPINIONS Progress in EGFR-mutant NSCLC: Where are we going? Transcript from a touchEXPERT OPINIONS Funded by an independent medical education request from AstraZeneca. This activity is provided by touchIME. www.touchoncology.com/cme-education/ 1 THE EXPERTS INTRODUCTION Watch leading non-small cell lung cancer (NSCLC) experts discuss the current standard and the latest advancements in epithelial growth factor receptor (EGFR)-targeted therapy. LEARNING OBJECTIVES After watching this touchEXPERT OPINIONS, DR RAFFAELE CALIFANO you should be better able to: The Christie Hospital, Manchester University Hospital University of Manchester, Manchester, UK • Describe the rationale for and clinical trial data associated with combination therapy in first-line EGFR-mutant NSCLC • Describe the mechanisms of resistance to third-generation EGFR-TKIs and strategies to manage drug resistance in the • second-line setting • Recall data on emerging therapeutic strategies for early-stage EGFR-mutant DR LECIA SEQUIST NSCLC and how this may impact the Harvard Medical School, Massachusetts metastatic treatment landscape General Hospital, Boston, MA, USA TOPICS DISCUSSED • Are combination strategies the way forward for EGFR-mutant NSCLC? • How will acquired resistance drive treatment choices in EGFR-mutant NSCLC? PROF. YI-LONG WU Guangdong Lung Cancer Institute, Guangdong • How do recent data in early-stage NSCLC Provincial People’s Hospital, Guangzhou, China impact the current treatment pathway for EGFR-mutant NSCLC? Launch date: 07 December 2020 www.touchoncology.com/cme-education/ 2 touchEXPERT OPINIONS ARE COMBINATION STRATEGIES THE WAY demonstrated that osimertinib determined a longer progression-free survival, but also a longer overall FORWARD FOR EGFR-MUTANT NSCLC? survival compared to first-generation EGFR TKIs. -

Kinase Drug Discovery 20 Years After Imatinib: Progress and Future Directions
REVIEWS Kinase drug discovery 20 years after imatinib: progress and future directions Philip Cohen 1 ✉ , Darren Cross 2 ✉ and Pasi A. Jänne 3 ✉ Abstract | Protein kinases regulate nearly all aspects of cell life, and alterations in their expression, or mutations in their genes, cause cancer and other diseases. Here, we review the remarkable progress made over the past 20 years in improving the potency and specificity of small-molecule inhibitors of protein and lipid kinases, resulting in the approval of more than 70 new drugs since imatinib was approved in 2001. These compounds have had a significant impact on the way in which we now treat cancers and non- cancerous conditions. We discuss how the challenge of drug resistance to kinase inhibitors is being met and the future of kinase drug discovery. Protein kinases In 2001, the first kinase inhibitor, imatinib, received FDA entered clinical trials in 1998, changed the perception Enzymes that catalyse transfer approval, providing the catalyst for an article with the of protein kinases as drug targets, which had previously of the γ- phosphate of ATP provocative title ‘Protein kinases — the major drug tar- received scepticism from many pharmaceutical com- to amino acid side chains in gets of the twenty- first century?’1. Imatinib inhibits the panies. Since then, hundreds of protein kinase inhibi- substrate proteins, such as serine, threonine and tyrosine Abelson (ABL) tyrosine kinase, which is expressed as a tors have been developed and tested in humans and, at residues. deregulated fusion protein, termed BCR–ABL, in nearly the time of writing, 76 have been approved for clinical all cases of chronic myeloid leukaemia (CML)2 and is use, mainly for the treatment of various cancers (FiG. -

(CHMP) Agenda for the Meeting on 17-20 May 2021 Chair: Harald Enzmann – Vice-Chair: Bruno Sepodes
17 May 2021 EMA/CHMP/276743/2021 Human Medicines Division Committee for medicinal products for human use (CHMP) Agenda for the meeting on 17-20 May 2021 Chair: Harald Enzmann – Vice-Chair: Bruno Sepodes 17 May 2021, 09:00 – 19:30, virtual meeting/ room 1C 18 May 2021, 08:30 – 19:30, virtual meeting/ room 1C 19 May 2021, 08:30 – 19:30, virtual meeting/ room 1C 20 May 2021, 08:30 – 19:30, virtual meeting/ room 1C Disclaimers Some of the information contained in this agenda is considered commercially confidential or sensitive and therefore not disclosed. With regard to intended therapeutic indications or procedure scopes listed against products, it must be noted that these may not reflect the full wording proposed by applicants and may also vary during the course of the review. Additional details on some of these procedures will be published in the CHMP meeting highlights once the procedures are finalised and start of referrals will also be available. Of note, this agenda is a working document primarily designed for CHMP members and the work the Committee undertakes. Note on access to documents Some documents mentioned in the agenda cannot be released at present following a request for access to documents within the framework of Regulation (EC) No 1049/2001 as they are subject to on- going procedures for which a final decision has not yet been adopted. They will become public when adopted or considered public according to the principles stated in the Agency policy on access to documents (EMA/127362/2006). Official address Domenico Scarlattilaan 6 ● 1083 HS Amsterdam ● The Netherlands Address for visits and deliveries Refer to www.ema.europa.eu/how-to-find-us Send us a question Go to www.ema.europa.eu/contact Telephone +31 (0)88 781 6000 An agency of the European Union © European Medicines Agency, 2021.