In This Issue
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Oncology Agents
APPROVED DRAFT PA Criteria Initial Approval Date: July 11, 2018 Revised Dates: January 20, 2021, October 14, 2020, October 10, 2018 CRITERIA FOR PRIOR AUTHORIZATION OncologyChemotherapy Agents BILLING CODE TYPE For drug coverage and provider type information, see the KMAP Reference Codes webpage. MANUAL GUIDELINES Prior authorization will be required for all current and future dose forms available. All medication-specific criteria will be reviewed according to the criteria below. Brand Name Generic Name Brand Name Generic Name Adcetris (brentuximab vedotin) Ibrance (palbociclib) Afinitor (everolimus) Iclusig (ponatinib hcl) Alecensa (alectinib hcl) Idhifa (enasidenib) Alunbrig (brigatinib) Imbruvica (ibrutinib) Arranon (nelarabine) Imfinzi (durvalumab) Avastin (bevacizumab) Inlyta (axitinib) Ayvakit (avapritinib) Ixempra (ixabepilone) Balversa (erdafitinib) Jakafi (ruxolitinib phosphate) Bavencio (avelumab) Jevtana (cabazitaxel) Belrapzo (bendamustine) Kadcyla (ado-trastuzumab) Bicnu (carmustine) Kanjinti (trastuzumab) Blincyto (blinatumomab) Keytruda (pembrolizumab) Bosulif (bosutinib) Kisqali (ribociclib) Braftovi (encorafenib) Kisqali Femara (ribociclib-letrozole) Brukinsa (zanubrutinib) Kyprolis (carfilzomib) Cabometyx (cabozantinib) Lartruvo (olaratumab) Calquence (acalabrutinib) Lenvima (lenvatinib) Cotellic (cobimetinib) Lonsurf (trifluridine-tipiracil) Cyramza (ramucirumab) Lorbrena (lorlatinib) Darzalex (daratumumab) Lynparza (olaparib) Darzalex Faspro (daratumumab and Matulane (procarbazine) hyaluronidase) Mekinist (trametinib) -

Fam-Trastuzumab Deruxtecan-Nxki Submitted by Daiichi
Dan Liang, PharmD Associate Director, Medical Information & Education Daiichi Sankyo, Inc. 211 Mount Airy Road Basking Ridge, NJ 07920 Phone: 908-992-7054 Email: [email protected] Date of request: July 9, 2020 NCCN Panel: Colon Cancer and Rectal Cancer On behalf of Daiichi Sankyo, Inc. and AstraZeneca Pharmaceuticals LP, I respectfully request the NCCN Guideline Panel for Colon and Rectal Cancers to review data from the clinical study1 in support of fam-trastuzumab deruxtecan-nxki, also known as T-DXd, as a monotherapy option for the treatment of patients with HER2-positive unresectable and/or metastatic colorectal cancer. Specific Changes: We respectfully ask the NCCN panel to consider the following: • COL-D1 through COL-D6 and REC-F1 through REC-F6, “Continuum of Care - Systemic Therapy for Advanced or Metastatic Disease” o Add “Fam-trastuzumab deruxtecan-nxki (HER2-positive and RAS and BRAF WT)” to the following: . COL-D1 and REC-F1: “Patient not appropriate for intensive therapy” . COL-D2 and REC-F2: “Previous oxaliplatin-based therapy without irinotecan” . COL-D3 and REC-F3: “Previous irinotecan-based therapy without oxaliplatin” . COL-D4 and REC-F4: “Previous treatment with oxaliplatin and irinotecan” . COL-D5 and REC-F5: “Previous therapy without irinotecan or oxaliplatin” . COL-D6 and REC-F6: “FOLFOX or CAPEOX or (FOLFOX or CAPEOX) + bevacizumab” • COL-D11 and REC-F11, “Systemic Therapy for Advanced or Metastatic Disease – Chemotherapy Regimens” o Add “Fam-trastuzumab deruxtecan-nxki 6.4 mg/kg IV on Day 1, cycled every 21 days” with a footnote: “fam-trastuzumab deruxtecan-nxki is approved for metastatic HER2-positive breast cancer at a different dose of 5.4 mg/kg IV on Day 1, cycled every 21 days” FDA Clearance: ENHERTU (fam-trastuzumab deruxtecan-nxki) is a HER2-directed antibody and topoisomerase inhibitor conjugate indicated for the treatment of adult patients with unresectable or metastatic HER2-positive breast cancer who have received two or more prior anti-HER2-based regimens in the metastatic setting. -

Targeting the Epidermal Growth Factor Receptor in EGFR-Mutated Lung Cancer: Current and Emerging Therapies
cancers Review Targeting the Epidermal Growth Factor Receptor in EGFR-Mutated Lung Cancer: Current and Emerging Therapies Karam Khaddour 1,*, Sushma Jonna 1, Alexander Deneka 2 , Jyoti D. Patel 3, Mohamed E. Abazeed 4, Erica Golemis 2 , Hossein Borghaei 5 and Yanis Boumber 3,6,* 1 Division of Hematology and Oncology, University of Illinois at Chicago, Chicago, IL 60612, USA; [email protected] 2 Fox Chase Cancer Center, Program in Molecular Therapeutics, Philadelphia, PA 19111, USA; [email protected] (A.D.); [email protected] (E.G.) 3 Robert H. Lurie Comprehensive Cancer Center, Division of Hematology/Oncology, Feinberg School of Medicine, Northwestern University, Chicago, IL 60611, USA; [email protected] 4 Robert H. Lurie Comprehensive Cancer Center, Department of Radiation Oncology, Feinberg School of Medicine, Northwestern University, Chicago, IL 60611, USA; [email protected] 5 Fox Chase Cancer Center, Department of Hematology and Oncology, Philadelphia, PA 19111, USA; [email protected] 6 Institute of Fundamental Medicine and Biology, Kazan Federal University, 420008 Kazan, Russia * Correspondence: [email protected] (K.K.); [email protected] (Y.B.) Simple Summary: Epidermal growth factor receptor (EGFR) mutations occur in a significant number Citation: Khaddour, K.; Jonna, S.; of lung cancer patients. Treatment outcomes in this subset of patients has greatly improved over the Deneka, A.; Patel, J.D.; Abazeed, M.E.; last decade after the introduction of EGFR tyrosine kinase inhibitors (TKIs), which demonstrated high Golemis, E.; Borghaei, H.; Boumber, Y. efficacy and improved survival in randomized clinical trials. Although EGFR TKIs became the stan- Targeting the Epidermal Growth dard of care in patients with EGFR-mutated lung cancer, resistance almost inevitably develops. -

Advances in Epidermal Growth Factor Receptor Specific Immunotherapy: Lessons to Be Learned from Armed Antibodies
www.oncotarget.com Oncotarget, 2020, Vol. 11, (No. 38), pp: 3531-3557 Review Advances in epidermal growth factor receptor specific immunotherapy: lessons to be learned from armed antibodies Fleury Augustin Nsole Biteghe1,*, Neelakshi Mungra2,*, Nyangone Ekome Toung Chalomie4, Jean De La Croix Ndong5, Jean Engohang-Ndong6, Guillaume Vignaux7, Eden Padayachee8, Krupa Naran2,* and Stefan Barth2,3,* 1Department of Radiation Oncology and Biomedical Sciences, Cedars-Sinai Medical, Los Angeles, CA, USA 2Medical Biotechnology & Immunotherapy Research Unit, Institute of Infectious Disease and Molecular Medicine, Faculty of Health Sciences, University of Cape Town, Cape Town, South Africa 3South African Research Chair in Cancer Biotechnology, Department of Integrative Biomedical Sciences, Faculty of Health Sciences, University of Cape Town, Cape Town, South Africa 4Sun Yat-Sen University, Zhongshan Medical School, Guangzhou, China 5Department of Orthopedic Surgery, New York University School of Medicine, New York, NY, USA 6Department of Biological Sciences, Kent State University at Tuscarawas, New Philadelphia, OH, USA 7Arctic Slope Regional Corporation Federal, Beltsville, MD, USA 8Department of Physiology, University of Kentucky, Lexington, KY, USA *These authors contributed equally to this work Correspondence to: Stefan Barth, email: [email protected] Keywords: epidermal growth factor receptor (EGFR); recombinant immunotoxins (ITs); targeted human cytolytic fusion proteins (hCFPs); recombinant antibody-drug conjugates (rADCs); recombinant antibody photoimmunoconjugates (rAPCs) Received: May 30, 2020 Accepted: August 11, 2020 Published: September 22, 2020 Copyright: © 2020 Biteghe et al. This is an open access article distributed under the terms of the Creative Commons Attribution License (CC BY 3.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. -

Interim Report for the First Quarter of 2020
Genmab Announces Financial Results for the First Quarter of 2020 May 6, 2020; Copenhagen, Denmark; Interim Report for the First Quarter Ended March 31, 2020 Highlights DARZALEX® (daratumumab) net sales increased approximately 49% compared to the first quarter of 2019 to USD 937 million, resulting in royalty income of DKK 775 million DARZALEX approved in Europe in combination with bortezomib, thalidomide and dexamethasone for the treatment of adult patients with newly diagnosed multiple myeloma who are eligible for autologous stem cell transplant U.S. FDA approved TEPEZZA™ (teprotumumab-trbw), developed and commercialized by Horizon Therapeutics, for thyroid eye disease U.S. FDA accepted, with priority review, Novartis’ supplemental Biologics License Application for subcutaneous ofatumumab in relapsing multiple sclerosis Anthony Pagano appointed Chief Financial Officer Anthony Mancini appointed Chief Operating Officer “Despite the unprecedented challenges posed by the coronavirus (COVID-19) pandemic, we will continue to invest in our innovative proprietary products, technologies and capabilities and use our world-class expertise in antibody drug development to create truly differentiated products with the potential to help cancer patients. While Genmab is closely monitoring the developments in the rapidly evolving landscape, we are extremely fortunate to have a solid financial foundation and a fabulous and committed team to carry us through these uncertain times,” said Jan van de Winkel, Ph.D., Chief Executive Officer of Genmab. Financial Performance First Quarter of 2020 Revenue was DKK 892 million in the first quarter of 2020 compared to DKK 591 million in the first quarter of 2019. The increase of DKK 301 million, or 51%, was mainly driven by higher DARZALEX royalties. -

Monoclonal Antibodies
MONOCLONAL ANTIBODIES ALEMTUZUMAB ® (CAMPATH 1H ) I. MECHANISM OF ACTION Antibody-dependent lysis of leukemic cells following cell surface binding. Alemtuzumab is a recombinant DNA-derived humanized monoclonal antibody that is directed against surface glycoprotein CD52. CD52 is expressed on the surface of normal and malignant B and T lymphocytes, NK cells, monocytes, macrophages, a subpopulation of granulocytes, and tissues of the male reproductive system (CD 52 is not expressed on erythrocytes or hematopoietic stem cells). The alemtuzumab antibody is an IgG1 kappa with human variable framework and constant regions, and complementarity-determining regions from a murine monoclonal antibody (campath 1G). II. PHARMACOKINETICS Cmax and AUC show dose proportionality over increasing dose ranges. The overall average half-life is 12 days. Peak and trough levels of Campath rise during the first weeks of Campath therapy, and approach steady state by week 6. The rise in serum Campath concentration corresponds with the reduction in malignant lymphocytes. III. DOSAGE AND ADMINISTRATION Campath can be administered intravenously or subcutaneously. Intravenous: Alemtuzumab therapy should be initiated at a dose of 3 mg administered as a 2-hour IV infusion daily. When the 3 mg dose is tolerated (i.e., ≤ Grade 2 infusion related side effects), the daily dose should be escalated to 10mg and continued until tolerated (i.e., ≤ Grade 2 infusion related side effects). When the 10 mg dose is tolerated, the maintenance dose of 30 mg may be initiated. The maintenance dose of alemtuzumab is 30 mg/day administered three times a week on alternate days (i.e. Monday, Wednesday, and Friday), for up to 12 weeks. -

Cyramza® (Ramucirumab)
Cyramza® (ramucirumab) (Intravenous) -E- Document Number: MODA-0405 Last Review Date: 07/01/2021 Date of Origin: 09/03/2019 Dates Reviewed: 09/2019, 10/2019, 01/2020, 04/2020, 07/2020, 10/2020, 01/2021, 04/2021, 07/2021 I. Length of Authorization Coverage will be provided for 6 months and may be renewed. II. Dosing Limits A. Quantity Limit (max daily dose) [NDC Unit]: • Cyramza 100 mg/10 mL: 4 vials per 14 days • Cyramza 500 mg/50 mL: 2 vials per 14 days B. Max Units (per dose and over time) [HCPCS Unit]: Gastric, Gastroesophageal, HCC, and Colorectal Cancer: • 180 billable units every 14 days NSCLC: • 240 billable units every 14 days III. Initial Approval Criteria 1 Coverage is provided in the following conditions: • Patient is at least 18 years of age; AND Universal Criteria 1 • Patient does not have uncontrolled severe hypertension; AND • Patient must not have had a surgical procedure within the preceding 28 days or have a surgical wound that has not fully healed; AND Gastric, Esophageal, and Gastro-esophageal Junction Adenocarcinoma † Ф 1-3,5-7,14,17,2e,5e • Used as subsequent therapy after fluoropyrimidine- or platinum-containing chemotherapy; AND • Used as a single agent OR in combination with paclitaxel; AND o Used for one of the following: Moda Health Plan, Inc. Medical Necessity Criteria Page 1/27 Proprietary & Confidential © 2021 Magellan Health, Inc. – Patient has unresectable locally advanced, recurrent, or metastatic disease; OR – Used as palliative therapy for locoregional disease in patients who are not surgical candidates -
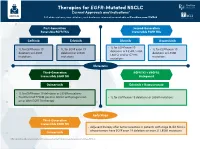
Therapies for EGFR-Mutated NSCLC Current Approvals and Indications1 Full Abbreviations, Accreditation, and Disclosure Information Available at Peerview.Com/CWE40
Therapies for EGFR-Mutated NSCLC 1 Current Approvals and Indications Full abbreviations, accreditation, and disclosure information available at PeerView.com/CWE40 First-Generation Second-Generation Reversible EGFR TKIs Irreversible EGFR TKIs Getinib Erlotinib Afatinib Dacomitinib • 1L for EGFR exon 19 • 1L for EGFR exon 19 • 1L for EGFR exon 19 • 1L for EGFR exon 19 deletions or L858R, S768I, deletions or L858R deletions or L858R deletions or L858R L861Q, and/or G719X mutations mutations mutations mutations Metastatic Third-Generation EGFR TKI + VEGFR2 Irreversible EGFR TKI Antagonist Osimertinib Erlotinib + Ramucirumab • 1L for EGFR exon 19 deletions or L858R mutations • Treatment of T790M-positive NSCLC with progression • 1L for EGFR exon 19 deletions or L858R mutations on or after EGFR TKI therapy Early Stage Third-Generation Irreversible EGFR TKI • Adjuvant therapy after tumor resection in patients with stage IB-IIIA NSCLC whose tumors have EGFR exon 19 deletions or exon 21 L858R mutations Osimertinib 1. https://www.fda.gov/drugs/resources-information-approved-drugs/hematologyoncology-cancer-approvals-safety-notifications. Molecular Testing Guidelines for NSCLC Latest Updates, Best Practices, and Patient-Reported Insights1 Full abbreviations, accreditation, and disclosure information available at PeerView.com/CWE40 Why Test Lung Cancer Patients for Genomic Alterations? • Genomic alterations are common in nonsquamous NSCLC (approximately 50%) • Targeted therapies produce better treatment outcomes (eg, higher response rates, improved -

Antibodies to Watch in 2021 Hélène Kaplona and Janice M
MABS 2021, VOL. 13, NO. 1, e1860476 (34 pages) https://doi.org/10.1080/19420862.2020.1860476 PERSPECTIVE Antibodies to watch in 2021 Hélène Kaplona and Janice M. Reichert b aInstitut De Recherches Internationales Servier, Translational Medicine Department, Suresnes, France; bThe Antibody Society, Inc., Framingham, MA, USA ABSTRACT ARTICLE HISTORY In this 12th annual installment of the Antibodies to Watch article series, we discuss key events in antibody Received 1 December 2020 therapeutics development that occurred in 2020 and forecast events that might occur in 2021. The Accepted 1 December 2020 coronavirus disease 2019 (COVID-19) pandemic posed an array of challenges and opportunities to the KEYWORDS healthcare system in 2020, and it will continue to do so in 2021. Remarkably, by late November 2020, two Antibody therapeutics; anti-SARS-CoV antibody products, bamlanivimab and the casirivimab and imdevimab cocktail, were cancer; COVID-19; Food and authorized for emergency use by the US Food and Drug Administration (FDA) and the repurposed Drug Administration; antibodies levilimab and itolizumab had been registered for emergency use as treatments for COVID-19 European Medicines Agency; in Russia and India, respectively. Despite the pandemic, 10 antibody therapeutics had been granted the immune-mediated disorders; first approval in the US or EU in 2020, as of November, and 2 more (tanezumab and margetuximab) may Sars-CoV-2 be granted approvals in December 2020.* In addition, prolgolimab and olokizumab had been granted first approvals in Russia and cetuximab saratolacan sodium was first approved in Japan. The number of approvals in 2021 may set a record, as marketing applications for 16 investigational antibody therapeutics are already undergoing regulatory review by either the FDA or the European Medicines Agency. -

Systemic Therapy Update
Systemic Therapy Update Volume 24 Issue 2 February 2021 For Health Professionals Who Care for Cancer Patients Inside This Issue: Editor’s Choice Revised Protocols, PPPOs and Patient Handouts COVID-19 Vaccine Guidance | New Programs: Ribociclib BR: BRAJACT, BRAJACTG, BRAJACTT, BRAJACTTG, BRAJACTW, BRAJCAP, and Fulvestrant for Advanced Breast Cancer BRAJLHRHAI, BRAJLHRHT, BRAJTTW, BRAVCAP, BRAVCLOD, BRAVEVEX, (UBRAVRBFLV) | Bevacizumab, Cisplatin and Paclitaxel for BRAVGEMT, BRAVLCAP, BRAVLHRHA, BRAVLHRHT, UBRAVPALAI, BRAVPTRAT, Gynecologic Malignancies (GOCISPBEV) | Gemtuzumab UBRAVRIBAI, BRAVTAX, BRAVTCAP, BRAVTW, BRLACTWAC, BRLATACG, BRLATWAC | GI: GIAAVCT, GIAJCAP, GIAJCAPOX, GIAJFFOX, GIAJRALOX, Ozogamicin with Chemotherapy for AML (ULKGEMOZ) | GIAVCAP, GIAVCAPB, GIAVCRT, GIAVTZCAP, GICAPIRI, GICAPOX, GICART, Bortezomib, Lenalidomide and Dexamethasone for GICIRB, GICOXB, GICPART, GIEFFOXRT, GIENACTRT, GIFFIRB, GIFFOXB, Untreated Multiple Myeloma (UMYBLDF) | Paclitaxel for UGIFFOXPAN, GIFIRINOX, GIFOLFOX, GIGAJCOX, GIGAJCPRT, GIGAJFFOX, Metastatic Angiosarcoma (SAAVTW) | Cemiplimab for GIGAVCC, GIGAVCCT, GIGAVCFT, GIGAVCOX, GIGAVCOXT, GIGAVFFOX, Cutaneous Squamous Cell Carcinoma (USMAVCEM) GIGAVFFOXT, GIGAVRAMT, GIGECC, GIGFLODOC, UGINETEV, GIPAJFIROX, GIPAJGCAP, GIPAVCAP, GIPNEVER, GIRAJCOX, GIRAJFFOX, GIRCAP, GIRCRT, Drug Update GIRINFRT | GO: GOCABR, GOCABRBEV, GOCISP, GOCXAJCAT, GOCXCAT, IV Famotidine to Replace IV Ranitidine GOCXCATB, GOCXCRT, GOENDCAT, GOOVBEVLD, GOOVBEVP, GOOVCATB, Medication Safety Corner GOOVCATM, GOOVCATR, -

Second-Line FOLFIRI Plus Ramucirumab with Or Without Prior
Cancer Chemotherapy and Pharmacology (2019) 84:307–313 https://doi.org/10.1007/s00280-019-03855-w ORIGINAL ARTICLE Second‑line FOLFIRI plus ramucirumab with or without prior bevacizumab for patients with metastatic colorectal cancer Takeshi Suzuki1,2 · Eiji Shinozaki1 · Hiroki Osumi1 · Izuma Nakayama1 · Yumiko Ota1 · Takashi Ichimura1 · Mariko Ogura1 · Takeru Wakatsuki1 · Akira Ooki1 · Daisuke Takahari1 · Mitsukuni Suenaga1 · Keisho Chin1 · Kensei Yamaguchi1 Received: 13 February 2019 / Accepted: 2 May 2019 / Published online: 7 May 2019 © Springer-Verlag GmbH Germany, part of Springer Nature 2019 Abstract Purpose Few data of folinic acid, fuorouracil, and irinotecan (FOLFIRI) plus ramucirumab (RAM) obtained in bevacizumab- naïve patients in clinical trials or routine clinical practice are available. The purpose of this retrospective study was to report the results of FOLFIRI plus RAM treatment as second-line chemotherapy for metastatic colorectal cancer (mCRC). Methods Seventy-four patients with mCRC who received second-line FOLFIRI + RAM mCRC therapy were stratifed by previous frst-line therapy to groups that had (PB) or had not (NPB) been given bevacizumab. The overall survival (OS), progression-free survival (PFS), and objective response were evaluated. Results The overall median PFS was 6.2 months (95% CI 4.6–9.3) and median OS was 17.0 months (95% CI 11.6–NA). Median PFS was 8.0 months (95% CI 4.9–11.2) in NPB patients and 5.0 months (95% CI 3.1–7.3) in PB patients (hazard ratio = 0.72, 95% CI 0.40–1.30, p = 0.28). The response rates were 23% and 3% in NPB and PB patients, respectively. -

PRESCRIBING INFORMATION These Highlights Do Not Include All the Information Needed to Use ------CONTRAINDICATIONS------VECTIBIX Safely and Effectively
HIGHLIGHTS OF PRESCRIBING INFORMATION These highlights do not include all the information needed to use -------------------------------CONTRAINDICATIONS------------------------------ VECTIBIX safely and effectively. See full prescribing information for None VECTIBIX. -----------------------WARNINGS AND PRECAUTIONS------------------------ VECTIBIX® (panitumumab) Injection for intravenous use Initial US Approval: 2006 • Dermatologic and Soft Tissue Toxicity: Monitor for dermatologic and soft tissue toxicities and withhold or discontinue Vectibix for severe or life-threatening complications. Limit sun exposure. (5.1, 5.7) WARNING: DERMATOLOGIC TOXICITY See full prescribing information for complete boxed warning. • Increased tumor progression, increased mortality, or lack of benefit in • Dermatologic toxicities were reported in 90% of patients and were patients with RAS-mutant mCRC. (2.1, 5.2) severe in 15% of patients receiving monotherapy. (2.3, 5.1, 6.1) • Electrolyte Depletion/Monitoring: Monitor electrolytes and institute appropriate treatment. (5.3) ----------------------------RECENT MAJOR CHANGES-------------------------- • Infusion Reactions: Terminate the infusion for severe infusion reactions. • Warnings and Precautions – Ocular Toxicities (5.8) 8/2021 (5.4) • Pulmonary Fibrosis/Interstitial Lung Disease (ILD): Permanently ----------------------------INDICATIONS AND USAGE--------------------------- discontinue Vectibix in patients developing ILD. (5.6) Vectibix is an epidermal growth factor receptor (EGFR) antagonist indicated •