A Reference Bacterial Genome Dataset Generated on the Miniontm Portable Single-Molecule Nanopore Sequencer Joshua Quick1,2, Aaron R Quinlan3 and Nicholas J Loman1*
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Gies for Increasing Citations
CORE Metadata, citation and similar papers at core.ac.uk Provided by International SERIES on Information Systems and Management in Creative eMedia (CreMedia) Maximized Research Impact: Effective Strate- gies for Increasing Citations Nader Ale Ebrahim*1, Hossein Gholizadeh2, Artur Lugmayr3 1Research Support Unit, Centre for Research Services, Institute of Research Management and Monitoring (IPPP), University of Malaya, Malaysia, Email: [email protected], *Corresponding author 2Department of Biomedical Engineering, Faculty of Engineering, University of Malaya, 50603 Kuala Lumpur, Malaysia 3VisLab., School of Media, Culture and Creative Arts, Curtin University, Perth, Australia Abstract The high competitive environment has forced higher education authori- ties to set their strategies to improve university ranking. Citations of published pa- pers are among the most widely used inputs to measure national and global uni- versity ranking (which accounts for 20% of QS, 30% of THE, and etc.). Therefore, from one hand, improving the citation impact of a search is one of the university manager’s strategies. On the other hand, the researchers are also looking for some helpful techniques to increase their citation record. This chapter by reviewing the relevant articles covers 48 different strategies for maximizing research impact and visibility. The results show that some features of article can help predict the num- ber of article views and citation counts. The findings presented in this chapter could be used by university authorities, authors, reviewers, and editors to maxim- ize the impact of articles in the scientific community. Keywords: University ranking, Improve citation, Citation frequency, Research impact, Open access, h-index Introduction The research output is an essential part of an institution’s measure and evaluation of research quality. -

Tracking the Popularity and Outcomes of All Biorxiv Preprints
1 Meta-Research: Tracking the popularity and outcomes of all bioRxiv 2 preprints 3 Richard J. Abdill1 and Ran Blekhman1,2 4 1 – Department of Genetics, Cell Biology, and Development, University of Minnesota, 5 Minneapolis, MN 6 2 – Department of Ecology, Evolution, and Behavior, University of Minnesota, St. Paul, 7 MN 8 9 ORCID iDs 10 RJA: 0000-0001-9565-5832 11 RB: 0000-0003-3218-613X 12 13 Correspondence 14 Ran Blekhman, PhD 15 University of Minnesota 16 MCB 6-126 17 420 Washington Avenue SE 18 Minneapolis, MN 55455 19 Email: [email protected] 1 20 Abstract 21 The growth of preprints in the life sciences has been reported widely and is 22 driving policy changes for journals and funders, but little quantitative information has 23 been published about preprint usage. Here, we report how we collected and analyzed 24 data on all 37,648 preprints uploaded to bioRxiv.org, the largest biology-focused preprint 25 server, in its first five years. The rate of preprint uploads to bioRxiv continues to grow 26 (exceeding 2,100 in October 2018), as does the number of downloads (1.1 million in 27 October 2018). We also find that two-thirds of preprints posted before 2017 were later 28 published in peer-reviewed journals, and find a relationship between the number of 29 downloads a preprint has received and the impact factor of the journal in which it is 30 published. We also describe Rxivist.org, a web application that provides multiple ways 31 to interact with preprint metadata. 32 Introduction 33 In the 30 days of September 2018, four leading biology journals – The Journal of 34 Biochemistry, PLOS Biology, Genetics and Cell – published 85 full-length research 35 articles. -

Open Science and the Role of Publishers in Reproducible Research
Open science and the role of publishers in reproducible research Authors: Iain Hrynaszkiewicz (Faculty of 1000), Peter Li (BGI), Scott Edmunds (BGI) Chapter summary Reproducible computational research is and will be facilitated by the wide availability of scientific data, literature and code which is freely accessible and, furthermore, licensed such that it can be reused, inteGrated and built upon to drive new scientific discoveries without leGal impediments. Scholarly publishers have an important role in encouraGing and mandating the availability of data and code accordinG to community norms and best practices, and developinG innovative mechanisms and platforms for sharinG and publishinG products of research, beyond papers in journals. Open access publishers, in particular the first commercial open access publisher BioMed Central, have played a key role in the development of policies on open access and open data, and increasing the use by scientists of leGal tools – licenses and waivers – which maximize reproducibility. Collaborations, between publishers and funders of scientific research, are vital for the successful implementation of reproducible research policies. The genomics and, latterly, other ‘omics communities historically have been leaders in the creation and wide adoption of policies on public availability of data. This has been throuGh policies, such as Fort Lauderdale and the Bermuda Principles; infrastructure, such as the INSDC databases; and incentives, such as conditions of journal publication. We review some of these policies and practices, and how these events relate to the open access publishinG movement. We describe the implementation and adoption of licenses and waivers prepared by Creative Commons, in science publishinG, with a focus on licensing of research data published in scholarly journals and data repositories. -

Citation Databases for Legal Scholarship
University at Buffalo School of Law Digital Commons @ University at Buffalo School of Law Law Librarian Journal Articles Law Librarian Scholarship 3-19-2020 Citation Databases for Legal Scholarship John R. Beatty University at Buffalo School of Law, [email protected] Follow this and additional works at: https://digitalcommons.law.buffalo.edu/law_librarian_articles Part of the Law Commons, Law Librarianship Commons, and the Scholarly Communication Commons Recommended Citation John R. Beatty, Citation Databases for Legal Scholarship, 39 Legal Ref. Servs. Q. 56 (2020). Available at: https://digitalcommons.law.buffalo.edu/law_librarian_articles/50 This is an Accepted Manuscript of an article published by Taylor & Francis in Legal Reference Services Quarterly on March 19, 2020, available online: https://www.tandfonline.com/doi/full/10.1080/0270319X.2020.1738198. This Article is brought to you for free and open access by the Law Librarian Scholarship at Digital Commons @ University at Buffalo School of Law. It has been accepted for inclusion in Law Librarian Journal Articles by an authorized administrator of Digital Commons @ University at Buffalo School of Law. For more information, please contact [email protected]. Citation Databases for Legal Scholarship John R. Beatty a* a Charles B. Sears Law Library, University at Buffalo, State University of New York, Buffalo, NY, USA John Beatty, Charles B. Sears Law Library, O’Brian Hall, Buffalo, NY 14260, [email protected], ORCID ID: 0000-0002-4940-8483 John R. Beatty is the Faculty Scholarship Outreach Librarian at the Charles B. Sears Law Library at the University at Buffalo. This is an Accepted Manuscript of an article published by Taylor & Francis in Legal Reference Services Quarterly on March 19, 2020, available online: https://www.tandfonline.com/doi/full/10.1080/0270319X.2020.1738198. -

Citations and Gamed Metrics: Academic Integrity Lost
Acad. Quest. (2021) 34.1 DOI 10.51845/34s.1.18 10.51845/34s.1.18 Articles Citations and Gamed Metrics: Academic Integrity Lost Jaime A. Teixeira da Silva Author-based metrics (ABMs) and journal-based metrics (JBMs), as well as citations, are the dominant currency of intellectual recognition in academia. These metrics and citations continue to be dominated by the Clarivate Analytics Journal Impact Factor (JIF) and are still widely used as a surrogate to determine prestige, scientific credit, and influence.1 However, their dominance in this area needs to be rethought, especially in China, which accounts for approximately 19 percent of total scientific research papers globally.2 China, like several other countries, games the JIF for financial compensation in pay-to-publish schemes based on JIF-carrying journals.3 A competing metric, CiteScore, is also showing signs of being gamed.4 It is possible that CiteScore may have evolved, following Goodhart’s Law, as JIF’s relevance as an “academic” metric began to fray. 5 The culture of metrics, especially the JIF, to measure success, reputation, and productivity, or JIF-based praise platforms such as Clarivate Analytics’ “Highly Cited Researchers” report inflate the problem because they elevate the perceived excellence of single individuals above all others, when in fact most publishing success is collaborative.6 The survival of many academics and journals in this publish-or-perish culture depends on such metrics, as authors seek journals with higher JIFs in which to publish their findings. In the gold 1 Erin C. McKiernan, Lesley A. Schimanski, Carol Muñoz Nieves, Lisa Matthias, Meredith T. -

Open Access—What's New, What's Worked, What Hasn't
Annual Meeting Reports Open Access—What’s New, What’s Worked, What Hasn’t Moderator: began talking about requiring OA, it was data and software be freely available and Deborah Kahn thought that publishers would jump right that reviews be signed. Executive Vice President, Publishing into the Gold OA model: An author pays The Journal of Neuroinflammation has BioMed Central a fee and an article appears in a journal been OA from its beginnings in 2004 London, United Kingdom in which all articles are OA. Not surpris- because the editors-in-chief believed that ingly, many publishers have been leery of OA was the wave of the future. Current Speakers: suddenly converting all their titles to Gold Editor-in-Chief Sue T Griffin present- Barbara Goldman OA without some idea of how it will affect ed the viewpoint of this established OA Director, Journals their revenue. Some are using a hybrid OA journal. Like GigaScience, the Journal of American Society for Microbiology model, in which an author can buy OA for Neuroinflammation has no embargo period, Washington, DC an article in a subscription journal. so all articles are made available on publi- Deborah Kahn, of BioMed Central, pub- cation. However, authors are charged a pro- Laurie Goodman lisher of more than 260 OA journals, led a cessing fee, whereas GigaScience has no fees Editor-in-Chief lively and informative panel that comprised for authors. Another difference between GigaScience five OA viewpoints: those of the author, these OA journals is in their style of peer BioMed Central the editor of an established OA journal, review. -

Assessing the Impact and Quality of Research Data Using Altmetrics and Other Indicators
Konkiel, S. (2020). Assessing the Impact and Quality of Research Data Using Altmetrics and Other Indicators. Scholarly Assessment Reports, 2(1): 13. DOI: https://doi.org/10.29024/sar.13 COMMISSIONED REPORT Assessing the Impact and Quality of Research Data Using Altmetrics and Other Indicators Stacy Konkiel Altmetric, US [email protected] Research data in all its diversity—instrument readouts, observations, images, texts, video and audio files, and so on—is the basis for most advancement in the sciences. Yet the assessment of most research pro- grammes happens at the publication level, and data has yet to be treated like a first-class research object. How can and should the research community use indicators to understand the quality and many poten- tial impacts of research data? In this article, we discuss the research into research data metrics, these metrics’ strengths and limitations with regard to formal evaluation practices, and the possible meanings of such indicators. We acknowledge the dearth of guidance for using altmetrics and other indicators when assessing the impact and quality of research data, and suggest heuristics for policymakers and evaluators interested in doing so, in the absence of formal governmental or disciplinary policies. Policy highlights • Research data is an important building block of scientific production, but efforts to develop a framework for assessing data’s impacts have had limited success to date. • Indicators like citations, altmetrics, usage statistics, and reuse metrics highlight the influence of research data upon other researchers and the public, to varying degrees. • In the absence of a shared definition of “quality”, varying metrics may be used to measure a dataset’s accuracy, currency, completeness, and consistency. -
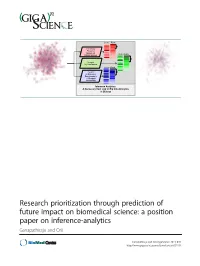
Research Prioritization Through Prediction of Future Impact on Biomedical Science: a Position Paper on Inference-Analytics Ganapathiraju and Orii
Score Rank Re-rank by Predicted Impact on Future Science Score Rank Re-rank by Confidence Score Rank Re-rank by Resource Requirements or Reagent Availability Inference Analytics: A Necessary Next step to Big Data Analytics in Biology Research prioritization through prediction of future impact on biomedical science: a position paper on inference-analytics Ganapathiraju and Orii Ganapathiraju and Orii GigaScience 2013, 2:11 http://www.gigasciencejournal.com/content/2/1/11 Ganapathiraju and Orii GigaScience 2013, 2:11 http://www.gigasciencejournal.com/content/2/1/11 RESEARCH Open Access Research prioritization through prediction of future impact on biomedical science: a position paper on inference-analytics Madhavi K Ganapathiraju1,2* and Naoki Orii1,2 Abstract Background: Advances in biotechnology have created “big-data” situations in molecular and cellular biology. Several sophisticated algorithms have been developed that process big data to generate hundreds of biomedical hypotheses (or predictions). The bottleneck to translating this large number of biological hypotheses is that each of them needs to be studied by experimentation for interpreting its functional significance. Even when the predictions are estimated to be very accurate, from a biologist’s perspective, the choice of which of these predictions is to be studied further is made based on factors like availability of reagents and resources and the possibility of formulating some reasonable hypothesis about its biological relevance. When viewed from a global perspective, say from that of a federal funding agency, ideally the choice of which prediction should be studied would be made based on which of them can make the most translational impact. -

A Locally Funded Puerto Rican Parrot (Amazona Vittata) Genome Sequencing Project Increases Avian Data and Advances Young Researcher Education Oleksyk Et Al
Downloaded from https://academic.oup.com/gigascience/article-abstract/1/1/2047-217X-1-14/2656136 by University of Lethbridge user on 20 March 2020 A locally funded Puerto Rican parrot (Amazona vittata) genome sequencing project increases avian data and advances young researcher education Oleksyk et al. Oleksyk et al. GigaScience 2012, 1:14 http://www.gigasciencejournal.com/content/1/1/14 28 Oleksyk et al. GigaScience 2012, 1:14 http://www.gigasciencejournal.com/content/1/1/14 DATANOTE Open Access A locally funded Puerto Rican parrot Downloaded from https://academic.oup.com/gigascience/article-abstract/1/1/2047-217X-1-14/2656136 by University of Lethbridge user on 20 March 2020 (Amazona vittata) genome sequencing project increases avian data and advances young researcher education Taras K Oleksyk1*, Jean-Francois Pombert2, Daniel Siu3, Anyimilehidi Mazo-Vargas1, Brian Ramos1, Wilfried Guiblet1, Yashira Afanador1, Christina T Ruiz-Rodriguez1,4, Michael L Nickerson4, David M Logue1, Michael Dean4, Luis Figueroa5, Ricardo Valentin6 and Juan-Carlos Martinez-Cruzado1 Abstract Background: Amazona vittata is a critically endangered Puerto Rican endemic bird, the only surviving native parrot species in the United States territory, and the first parrot in the large Neotropical genus Amazona, to be studied on a genomic scale. Findings: In a unique community-based funded project, DNA from an A. vittata female was sequenced using a HiSeq Illumina platform, resulting in a total of ~42.5 billion nucleotide bases. This provided approximately 26.89x average coverage depth at the completion of this funding phase. Filtering followed by assembly resulted in 259,423 contigs (N50 = 6,983 bp, longest = 75,003 bp), which was further scaffolded into 148,255 fragments (N50 = 19,470, longest = 206,462 bp). -

Efficient and Accurate Error Correction for Illumina RNA-Seq Reads Li Song1 and Liliana Florea1,2*
Song and Florea GigaScience (2015) 4:48 DOI 10.1186/s13742-015-0089-y TECHNICAL NOTE Open Access Rcorrector: efficient and accurate error correction for Illumina RNA-seq reads Li Song1 and Liliana Florea1,2* Abstract Background: Next-generation sequencing of cellular RNA (RNA-seq) is rapidly becoming the cornerstone of transcriptomic analysis. However, sequencing errors in the already short RNA-seq reads complicate bioinformatics analyses, in particular alignment and assembly. Error correction methods have been highly effective for whole-genome sequencing (WGS) reads, but are unsuitable for RNA-seq reads, owing to the variation in gene expression levels and alternative splicing. Findings: We developed a k-mer based method, Rcorrector, to correct random sequencing errors in Illumina RNA-seq reads. Rcorrector uses a De Bruijn graph to compactly represent all trusted k-mers in the input reads. Unlike WGS read correctors, which use a global threshold to determine trusted k-mers, Rcorrector computes a local threshold at every position in a read. Conclusions: Rcorrector has an accuracy higher than or comparable to existing methods, including the only other method (SEECER) designed for RNA-seq reads, and is more time and memory efficient. With a 5 GB memory footprint for 100 million reads, it can be run on virtually any desktop or server. The software is available free of charge under the GNU General Public License from https://github.com/mourisl/Rcorrector/. Keywords: Next-generation sequencing, RNA-seq, Error correction, k-mers Introduction based methods, which are the most popular of the Next-generation sequencing of cellular RNA (RNA-seq) three, classify a k-mer as trusted or untrusted depending has become the foundation of virtually every transcrip- on whether the number of occurrences in the input tomic analysis. -

Impact Factor Volatility to a Single Paper: a Comprehensive Analysis
Impact Factor volatility to a single paper: A comprehensive analysis Manolis Antonoyiannakisa,b aDepartment of Applied Physics & Applied Mathematics, Columbia University, 500 W. 120th St., Mudd 200, New York, NY 10027 bAmerican Physical Society, Editorial Office, 1 Research Road, Ridge, NY 11961-2701 ORCID: 0000-0001-6174-0668 Abstract We study how a single paper affects the Impact Factor (IF) by analyzing data from 3,088,511 papers published in 11639 journals in the 2017 Journal Citation Reports of Clarivate Analytics. We find that IFs are highly volatile. For example, the top-cited paper of 381 journals caused their IF to increase by more than 0.5 points, while for 818 journals the relative increase exceeded 25%. And one in 10 journals had their IF boosted by more than 50% by their top three cited papers. Because the single-paper effect on the IF is inversely proportional to journal size, small journals are rewarded much more strongly than large journals for a highly-cited paper, while they are pe- nalized more for a low-cited paper, especially if their IF is high. This skewed reward mechanism incentivizes high-IF journals to stay small, to remain competitive in rankings. We discuss the im- plications for breakthrough papers appearing in prestigious journals. We question the reliability of IF rankings given the high IF sensitivity to a few papers for thousands of journals. Keywords: Science of Science, Impact Factor, Volatility, Citation Distributions, Bibliostatistics 1. Introduction & Motivation The effect of a journal’s scale (i.e., size) on its citation average cannot be overstated. -

The 2019 Oxford Journals Collection
1 | OXFORD JOURNALS The 2019 Oxford Journals Collection 357 high-quality journals 17 journals added in 2019 Access to content back to 1996 academic.oup.com/journals/collection Medicine | Life Sciences | Humanities | Social Sciences Mathematics@OUPLibraries & Physical Sciences | Law For further information visit: 2 | OXFORD JOURNALS The 2019 Oxford Journals Collection published in partnership with many of the world’s Over 70% most influential scholarly and professional societies 357 Over 70% high-quality of journals have an SOCIAL Impact Factor journals SCIENCES HUMANITIES 6 Over 80% subject of these are in the LIFE collections SCIENCES MEDICINE top 50% of at least one category MATHEMATICS & PHYSICAL 6 SCIENCES LAW journals ranked #1 in at least one category Nobel Prize Over Joining in winning authors 2019… 100 in million journals in both all established and downloads 6 subject collections 17 emerging fields in 2017 Impact Factor and Ranking Source: 2017 Journal Citation Reports® (Clarivate Analytics, 2018) @OUPLibraries For further information visit: COLLECTION 2019 | 3 Rankings in Best JCR Category Medicine Life Sciences Humanities Social Sciences Top 10% Mathematics & Physical Sciences 10% to 25% 25% to 50% Bottom 50% Law No JCR Entry 0% 10% 20% 30% 40% 50% 60% 70% 80% 90% 100% Source: 2017 Journal Citation Reports® (Clarivate Analytics, 2018) Cost per Citation ($) Maths & GRAND Food Health Agriculture Astronomy Biology Botany Chemistry Geology Computer Physics Zoology TOTAL Science Sciences Science OUP 0.15 0.20 0.10 0.17 0.18 0.20 0.03 0.17 0.09 0.23 0.31 0.15 Survey 0.28 0.30 0.18 0.25 0.28 0.22 0.38 0.25 0.21 0.52 0.26 0.54 Business & Language Philosophy Political Social Education Geography History Law Music Psychology Sociology Economics & Literature & Religion Science Sciences OUP 0.22 0.47 0.52 0.83 0.60 1.15 0.50 0.60 0.52 0.14 0.25 0.22 Survey 0.60 0.72 0.45 1.63 1.92 0.52 3.14 6.95 0.75 0.27 0.88 0.55 The survey data shows the average cost per citation of journals in Clarivate Analytics Indexes (source: Library Journal Periodicals Price Survey 2018).