KCNK18 Biallelic Variants Associated with Intellectual Disability and Neurodevelopmental Disorders Alter TRESK Channel Activity
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Potassium Channels in Epilepsy
Downloaded from http://perspectivesinmedicine.cshlp.org/ on September 28, 2021 - Published by Cold Spring Harbor Laboratory Press Potassium Channels in Epilepsy Ru¨diger Ko¨hling and Jakob Wolfart Oscar Langendorff Institute of Physiology, University of Rostock, Rostock 18057, Germany Correspondence: [email protected] This review attempts to give a concise and up-to-date overview on the role of potassium channels in epilepsies. Their role can be defined from a genetic perspective, focusing on variants and de novo mutations identified in genetic studies or animal models with targeted, specific mutations in genes coding for a member of the large potassium channel family. In these genetic studies, a demonstrated functional link to hyperexcitability often remains elusive. However, their role can also be defined from a functional perspective, based on dy- namic, aggravating, or adaptive transcriptional and posttranslational alterations. In these cases, it often remains elusive whether the alteration is causal or merely incidental. With 80 potassium channel types, of which 10% are known to be associated with epilepsies (in humans) or a seizure phenotype (in animals), if genetically mutated, a comprehensive review is a challenging endeavor. This goal may seem all the more ambitious once the data on posttranslational alterations, found both in human tissue from epilepsy patients and in chronic or acute animal models, are included. We therefore summarize the literature, and expand only on key findings, particularly regarding functional alterations found in patient brain tissue and chronic animal models. INTRODUCTION TO POTASSIUM evolutionary appearance of voltage-gated so- CHANNELS dium (Nav)andcalcium (Cav)channels, Kchan- nels are further diversified in relation to their otassium (K) channels are related to epilepsy newer function, namely, keeping neuronal exci- Psyndromes on many different levels, ranging tation within limits (Anderson and Greenberg from direct control of neuronal excitability and 2001; Hille 2001). -

A Dominant-Negative Mutation in the TRESK Potassium Channel Is Linked to Familial Migraine with Aura
LETTERS A dominant-negative mutation in the TRESK potassium channel is linked to familial migraine with aura Ronald G Lafrenière1,2,13, M Zameel Cader3,4,13, Jean-François Poulin2, Isabelle Andres-Enguix5, Maryse Simoneau2, Namrata Gupta2, Karine Boisvert2, François Lafrenière2, Shannon McLaughlan2, Marie-Pierre Dubé6, Martin M Marcinkiewicz7, Sreeram Ramagopalan8, Olaf Ansorge9, Bernard Brais1, Jorge Sequeiros10, Jose Maria Pereira-Monteiro11, Lyn R Griffiths12, Stephen J Tucker5, George Ebers8 & Guy A Rouleau1,2 Migraine with aura is a common, debilitating, recurrent of the trigeminal ganglion afferents5 and may lead to central sensiti- headache disorder associated with transient and reversible focal zation. Considerable insights into the pathogenesis of migraine have neurological symptoms1. A role has been suggested for the come from the investigation of the rare autosomal dominant subtype two-pore domain (K2P) potassium channel, TWIK-related spinal of migraine with aura, familial hemiplegic migraine. Three suscepti- cord potassium channel (TRESK, encoded by KCNK18), in pain bility genes (CACNA1A, ATP1A2 and SCNA1), which encode either pathways and general anaesthesia2. We therefore examined ion channels or ion transport proteins, have so far been identified6–8, whether TRESK is involved in migraine by screening the and it is likely that mutations in these genes reduce the threshold for KCNK18 gene in subjects diagnosed with migraine. Here we CSD9,10. However, such mutations are not found in typical migraine report a frameshift mutation, F139WfsX24, which segregates with aura, suggesting that other ion channels may be involved. perfectly with typical migraine with aura in a large pedigree. K2P channels are expressed throughout the central nervous system We also identified prominent TRESK expression in migraine- and have a key role in controlling neuronal resting membrane potential salient areas such as the trigeminal ganglion. -

Distinguishing Pleiotropy from Linked QTL Between Milk Production Traits
Cai et al. Genet Sel Evol (2020) 52:19 https://doi.org/10.1186/s12711-020-00538-6 Genetics Selection Evolution RESEARCH ARTICLE Open Access Distinguishing pleiotropy from linked QTL between milk production traits and mastitis resistance in Nordic Holstein cattle Zexi Cai1*†, Magdalena Dusza2†, Bernt Guldbrandtsen1, Mogens Sandø Lund1 and Goutam Sahana1 Abstract Background: Production and health traits are central in cattle breeding. Advances in next-generation sequencing technologies and genotype imputation have increased the resolution of gene mapping based on genome-wide association studies (GWAS). Thus, numerous candidate genes that afect milk yield, milk composition, and mastitis resistance in dairy cattle are reported in the literature. Efect-bearing variants often afect multiple traits. Because the detection of overlapping quantitative trait loci (QTL) regions from single-trait GWAS is too inaccurate and subjective, multi-trait analysis is a better approach to detect pleiotropic efects of variants in candidate genes. However, large sample sizes are required to achieve sufcient power. Multi-trait meta-analysis is one approach to deal with this prob- lem. Thus, we performed two multi-trait meta-analyses, one for three milk production traits (milk yield, protein yield and fat yield), and one for milk yield and mastitis resistance. Results: For highly correlated traits, the power to detect pleiotropy was increased by multi-trait meta-analysis com- pared with the subjective assessment of overlapping of single-trait QTL confdence intervals. Pleiotropic efects of lead single nucleotide polymorphisms (SNPs) that were detected from the multi-trait meta-analysis were confrmed by bivariate association analysis. The previously reported pleiotropic efects of variants within the DGAT1 and MGST1 genes on three milk production traits, and pleiotropic efects of variants in GHR on milk yield and fat yield were con- frmed. -

Transcriptomic Analysis of Native Versus Cultured Human and Mouse Dorsal Root Ganglia Focused on Pharmacological Targets Short
bioRxiv preprint doi: https://doi.org/10.1101/766865; this version posted September 12, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. Transcriptomic analysis of native versus cultured human and mouse dorsal root ganglia focused on pharmacological targets Short title: Comparative transcriptomics of acutely dissected versus cultured DRGs Andi Wangzhou1, Lisa A. McIlvried2, Candler Paige1, Paulino Barragan-Iglesias1, Carolyn A. Guzman1, Gregory Dussor1, Pradipta R. Ray1,#, Robert W. Gereau IV2, # and Theodore J. Price1, # 1The University of Texas at Dallas, School of Behavioral and Brain Sciences and Center for Advanced Pain Studies, 800 W Campbell Rd. Richardson, TX, 75080, USA 2Washington University Pain Center and Department of Anesthesiology, Washington University School of Medicine # corresponding authors [email protected], [email protected] and [email protected] Funding: NIH grants T32DA007261 (LM); NS065926 and NS102161 (TJP); NS106953 and NS042595 (RWG). The authors declare no conflicts of interest Author Contributions Conceived of the Project: PRR, RWG IV and TJP Performed Experiments: AW, LAM, CP, PB-I Supervised Experiments: GD, RWG IV, TJP Analyzed Data: AW, LAM, CP, CAG, PRR Supervised Bioinformatics Analysis: PRR Drew Figures: AW, PRR Wrote and Edited Manuscript: AW, LAM, CP, GD, PRR, RWG IV, TJP All authors approved the final version of the manuscript. 1 bioRxiv preprint doi: https://doi.org/10.1101/766865; this version posted September 12, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. -

Ion Channels
UC Davis UC Davis Previously Published Works Title THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: Ion channels. Permalink https://escholarship.org/uc/item/1442g5hg Journal British journal of pharmacology, 176 Suppl 1(S1) ISSN 0007-1188 Authors Alexander, Stephen PH Mathie, Alistair Peters, John A et al. Publication Date 2019-12-01 DOI 10.1111/bph.14749 License https://creativecommons.org/licenses/by/4.0/ 4.0 Peer reviewed eScholarship.org Powered by the California Digital Library University of California S.P.H. Alexander et al. The Concise Guide to PHARMACOLOGY 2019/20: Ion channels. British Journal of Pharmacology (2019) 176, S142–S228 THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: Ion channels Stephen PH Alexander1 , Alistair Mathie2 ,JohnAPeters3 , Emma L Veale2 , Jörg Striessnig4 , Eamonn Kelly5, Jane F Armstrong6 , Elena Faccenda6 ,SimonDHarding6 ,AdamJPawson6 , Joanna L Sharman6 , Christopher Southan6 , Jamie A Davies6 and CGTP Collaborators 1School of Life Sciences, University of Nottingham Medical School, Nottingham, NG7 2UH, UK 2Medway School of Pharmacy, The Universities of Greenwich and Kent at Medway, Anson Building, Central Avenue, Chatham Maritime, Chatham, Kent, ME4 4TB, UK 3Neuroscience Division, Medical Education Institute, Ninewells Hospital and Medical School, University of Dundee, Dundee, DD1 9SY, UK 4Pharmacology and Toxicology, Institute of Pharmacy, University of Innsbruck, A-6020 Innsbruck, Austria 5School of Physiology, Pharmacology and Neuroscience, University of Bristol, Bristol, BS8 1TD, UK 6Centre for Discovery Brain Science, University of Edinburgh, Edinburgh, EH8 9XD, UK Abstract The Concise Guide to PHARMACOLOGY 2019/20 is the fourth in this series of biennial publications. The Concise Guide provides concise overviews of the key properties of nearly 1800 human drug targets with an emphasis on selective pharmacology (where available), plus links to the open access knowledgebase source of drug targets and their ligands (www.guidetopharmacology.org), which provides more detailed views of target and ligand properties. -

Genomics Analysis of Potassium Channel Genes in Songbirds Reveals
Lovell et al. BMC Genomics 2013, 14:470 http://www.biomedcentral.com/1471-2164/14/470 RESEARCH ARTICLE Open Access Genomics analysis of potassium channel genes in songbirds reveals molecular specializations of brain circuits for the maintenance and production of learned vocalizations Peter V Lovell, Julia B Carleton and Claudio V Mello* Abstract Background: A fundamental question in molecular neurobiology is how genes that determine basic neuronal properties shape the functional organization of brain circuits underlying complex learned behaviors. Given the growing availability of complete vertebrate genomes, comparative genomics represents a promising approach to address this question. Here we used genomics and molecular approaches to study how ion channel genes influence the properties of the brain circuitry that regulates birdsong, a learned vocal behavior with important similarities to human speech acquisition. We focused on potassium (K-)Channels, which are major determinants of neuronal cell excitability. Starting with the human gene set of K-Channels, we used cross-species mRNA/protein alignments, and syntenic analysis to define the full complement of orthologs, paralogs, allelic variants, as well as novel loci not previously predicted in the genome of zebra finch (Taeniopygia guttata). We also compared protein coding domains in chicken and zebra finch orthologs to identify genes under positive selective pressure, and those that contained lineage-specific insertions/deletions in functional domains. Finally, we conducted comprehensive in situ hybridizations to determine the extent of brain expression, and identify K-Channel gene enrichments in nuclei of the avian song system. Results: We identified 107 K-Channel finch genes, including 6 novel genes common to non-mammalian vertebrate lineages. -

Bioelectric-Calcineurin Signaling Module Regulates Allometric Growth and Size of the Zebrafsh Fn Received: 8 March 2018 Jacob M
www.nature.com/scientificreports OPEN Bioelectric-calcineurin signaling module regulates allometric growth and size of the zebrafsh fn Received: 8 March 2018 Jacob M. Daane1, Jennifer Lanni1,2, Ina Rothenberg3, Guiscard Seebohm3, Charles W. Higdon4, Accepted: 18 June 2018 Stephen L. Johnson4 & Matthew P. Harris1 Published: xx xx xxxx The establishment of relative size of organs and structures is paramount for attaining fnal form and function of an organism. Importantly, variation in the proportions of structures frequently underlies adaptive change in morphology in evolution and maybe a common mechanism underlying selection. However, the mechanism by which growth is integrated within tissues during development to achieve proper proportionality is poorly understood. We have shown that signaling by potassium channels mediates coordinated size regulation in zebrafsh fns. Recently, calcineurin inhibitors were shown to elicit changes in zebrafsh fn allometry as well. Here, we identify the potassium channel kcnk5b as a key player in integrating calcineurin’s growth efects, in part through regulation of the cytoplasmic C-terminus of the channel. We propose that the interaction between Kcnk5b and calcineurin acts as a signaling node to regulate allometric growth. Importantly, we fnd that this regulation is epistatic to inherent mechanisms instructing overall size as inhibition of calcineurin is able to bypass genetic instruction of size as seen in sof and wild-type fns, however, it is not sufcient to re-specify positional memory of size of the fn. These fndings integrate classic signaling mediators such as calcineurin with ion channel function in the regulation of size and proportion during growth. Te establishment of relative proportion of structures and organs is essential for the normal physiology and function of an organism. -

Genetics of Migraine - Is There Any Progress?
Journal of Neurology & Stroke Genetics of Migraine - Is There any Progress? Abstract Review Article Nowadays migraine ranks 9th in the list of leading causes of disability among Volume 7 Issue 4 - 2017 population. In Russia migraine prevalence is two times higher than the world one-century history of studying migraine, science until now cannot explain many index and inflicts a considerable damage on the state economy. Despite almost – the therapy of patients with migraine is not sufficiently effective. Today one cases of attack occurrence. It causes difficulties both for diagnosis and treatment of the investigation directions is searching of migraine biomarkers confirming diagnosis. In this review we attempted to generalize the results of available works 1Faculty of Biology of Lomonosov Moscow State University, Keywords:targeted at searching genetic markers of migraine. Russia 2University diagnostic laboratory, Russia Migraine; Gene; Polymorphism 3University Headache Clinic, Russia 4Department of Neuroscience, I.M.Sechenov First Moscow State Medical University, Russia Introduction 5Centre of Theoretical Problems of Physico-Chemical Pharmacology, Russia 6I.I. Mechnikov Research Institute for Vaccines and Sera RAMS, Russia Migraine is now one of the leading causes of disability (ranks 7Department of neurology and neurosurgery, I.M. Sechenov 9th according to the WHO), comparable to such diseases as First Moscow State Medical University, Russia cancer, diabetes, cardiovascular diseases and others. In the female *Corresponding author: prevalencepopulation, inmigraine-related the world for 1 disability year in the ratio adult promotes population this disease ranges to 3rd place. According to epidemiological studies, migraine Eugene Klimov, Faculty of Biology of Lomonosov Moscow State University, Russia, Email: on average from 10.2% [1] to 14.7 % [2]. -

Robles JTO Supplemental Digital Content 1
Supplementary Materials An Integrated Prognostic Classifier for Stage I Lung Adenocarcinoma based on mRNA, microRNA and DNA Methylation Biomarkers Ana I. Robles1, Eri Arai2, Ewy A. Mathé1, Hirokazu Okayama1, Aaron Schetter1, Derek Brown1, David Petersen3, Elise D. Bowman1, Rintaro Noro1, Judith A. Welsh1, Daniel C. Edelman3, Holly S. Stevenson3, Yonghong Wang3, Naoto Tsuchiya4, Takashi Kohno4, Vidar Skaug5, Steen Mollerup5, Aage Haugen5, Paul S. Meltzer3, Jun Yokota6, Yae Kanai2 and Curtis C. Harris1 Affiliations: 1Laboratory of Human Carcinogenesis, NCI-CCR, National Institutes of Health, Bethesda, MD 20892, USA. 2Division of Molecular Pathology, National Cancer Center Research Institute, Tokyo 104-0045, Japan. 3Genetics Branch, NCI-CCR, National Institutes of Health, Bethesda, MD 20892, USA. 4Division of Genome Biology, National Cancer Center Research Institute, Tokyo 104-0045, Japan. 5Department of Chemical and Biological Working Environment, National Institute of Occupational Health, NO-0033 Oslo, Norway. 6Genomics and Epigenomics of Cancer Prediction Program, Institute of Predictive and Personalized Medicine of Cancer (IMPPC), 08916 Badalona (Barcelona), Spain. List of Supplementary Materials Supplementary Materials and Methods Fig. S1. Hierarchical clustering of based on CpG sites differentially-methylated in Stage I ADC compared to non-tumor adjacent tissues. Fig. S2. Confirmatory pyrosequencing analysis of DNA methylation at the HOXA9 locus in Stage I ADC from a subset of the NCI microarray cohort. 1 Fig. S3. Methylation Beta-values for HOXA9 probe cg26521404 in Stage I ADC samples from Japan. Fig. S4. Kaplan-Meier analysis of HOXA9 promoter methylation in a published cohort of Stage I lung ADC (J Clin Oncol 2013;31(32):4140-7). Fig. S5. Kaplan-Meier analysis of a combined prognostic biomarker in Stage I lung ADC. -

Potassium Channels and Their Potential Roles in Substance Use Disorders
International Journal of Molecular Sciences Review Potassium Channels and Their Potential Roles in Substance Use Disorders Michael T. McCoy † , Subramaniam Jayanthi † and Jean Lud Cadet * Molecular Neuropsychiatry Research Branch, NIDA Intramural Research Program, Baltimore, MD 21224, USA; [email protected] (M.T.M.); [email protected] (S.J.) * Correspondence: [email protected]; Tel.: +1-443-740-2656 † Equal contributions (joint first authors). Abstract: Substance use disorders (SUDs) are ubiquitous throughout the world. However, much re- mains to be done to develop pharmacotherapies that are very efficacious because the focus has been mostly on using dopaminergic agents or opioid agonists. Herein we discuss the potential of using potassium channel activators in SUD treatment because evidence has accumulated to support a role of these channels in the effects of rewarding drugs. Potassium channels regulate neuronal action potential via effects on threshold, burst firing, and firing frequency. They are located in brain regions identified as important for the behavioral responses to rewarding drugs. In addition, their ex- pression profiles are influenced by administration of rewarding substances. Genetic studies have also implicated variants in genes that encode potassium channels. Importantly, administration of potassium agonists have been shown to reduce alcohol intake and to augment the behavioral effects of opioid drugs. Potassium channel expression is also increased in animals with reduced intake of methamphetamine. Together, these results support the idea of further investing in studies that focus on elucidating the role of potassium channels as targets for therapeutic interventions against SUDs. Keywords: alcohol; cocaine; methamphetamine; opioids; pharmacotherapy Citation: McCoy, M.T.; Jayanthi, S.; Cadet, J.L. -
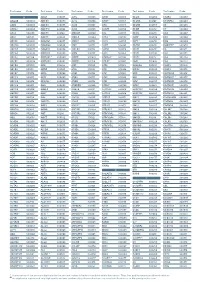
Aagab S00002 Aars S00003 Aars2 S00004 Aass S02483
Test name Code Test name Code Test name Code Test name Code Test name Code Test name Code A ADAR S00053 ALPL S00105 ARSB S00153 BCL10 S02266 C5AR2 S00263 AAGAB S00002 ADCK3 S00054 ALS2 S00106 ARSE * S00154 BCL11A S02167 C5ORF42 S00264 AARS S00003 ADCK4 S00055 ALX3 S00107 ARX S00155 BCL11B S02358 C6 S00265 AARS2 S00004 ADCY10 S02094 ALX4 S00108 ASAH1 S00156 BCOR S00212 C7 S00266 AASS S02483 ADCY3 S02184 AMACR S00109 ASL S00157 BCS1L S00213 C8A S00267 ABAT S02191 ADCY5 S02226 AMELX S02289 ASNS * S02508 BDNF S02509 C8B S00268 ABCA1 S00005 ADGRG1 S00057 AMER1 S00110 ASPA S00158 BDP1 * S00214 C8G S00269 ABCA12 S00006 ADGRG6 S02548 AMH S00111 ASPH S02425 BEAN1 S00215 C8ORF37 S00270 ABCA3 S00007 ADGRV1 S00058 AMHR2 S00112 ASPM S00159 BEST1 S00216 C9 S00271 ABCA4 S00008 ADIPOQ S00059 AMN S00113 ASS1 S00160 BFSP1 S02280 CA2 S00272 ABCA7 S02106 ADIPOR1 * S00060 AMPD1 S02670 ATAD3A * S02196 BFSP2 S00217 CA4 S02303 ABCB11 S00009 ADIPOR2 S00061 AMPD2 S02128 ATCAY S00162 BGN S02633 CA8 S00273 ABCB4 S00010 ADK S02595 AMT S00114 ATF6 S00163 BHLHA9 S00218 CABP2 S00274 ABCB6 S00011 ADNP S02320 ANG S00115 ATIC S02458 BICD2 S00220 CABP4 S00275 ABCB7 S00012 ADSL S00062 ANK1 S00116 ATL1 S00164 BIN1 S00221 CACNA1A S00276 ABCC2 S00013 AFF2 S00063 ANK2 S00117 ATL3 S00165 BLK S00222 CACNA1C * S00277 ABCC6 * S00014 AFG3L2 * S00064 ANKH S00118 ATM S00166 BLM S00223 CACNA1D S00278 ABCC8 S00015 AGA S00065 ANKRD11 * S02140 ATOH7 S02390 BLNK S02281 CACNA1F S00279 ABCC9 S00016 AGBL5 S02452 ANKS6 S00121 ATP13A2 S00168 BLOC1S3 S00224 CACNA1H S00280 ABCD1 * S00017 AGK * -

Viewed Papers: Yin K, Baillie GJ and Vetter I
A Pharmacological and Transcriptomic approach to exploring Novel Pain Targets Kathleen Yin Bachelor of Pharmacy A thesis submitted for the degree of Doctor of Philosophy at The University of Queensland in 2016 Institute for Molecular Bioscience i Abstract Ever since the discovery that mutations in the voltage-gated sodium channel 1.7 protein are responsible for human congenital insensitivity to pain, the voltage-gated sodium channel (NaV) family of ion channels has been the subject of intense research with the hope of discovering novel analgesics. We now know that NaV1.7 deletion in select neuronal populations yield different phenotypes, with the deletion of NaV1.7 in all sensory neurons being successful at abolishing mechanical and heat-induced pain. However, it is rapidly becoming apparent that a number of pain syndromes are not modulated by NaV1.7 at all, such as oxaliplatin-mediated neuropathy. It is particularly interesting to note that the loss of NaV1.7 function is also associated with the selective inhibition of pain mediated by specific stimuli, such as that observed in burn-induced pain where NaV1.7 gene knockout abolished thermal allodynia but did not affect mechanical allodynia. Accordingly, significant interests exist in delineating the contribution of other NaV isoforms in modality-specific pain pathways. Two other isoforms, NaV1.6 and NaV1.8, are now specifically implicated in some NaV1.7-independent conditions such as oxaliplatin-induced cold allodynia. Such selective contributions of specific ion channel isoforms to pain highlight the need to discover other putative protein targets involved in mediating nociception. The aim of my work is therefore to discover selective molecular inhibitors of NaV1.6 and NaV1.8, to find useful cell models for peripheral nociceptors, to investigate the roles of NaV1.6 and NaV1.8 in an animal model of burn-induced pain, and to screen for putative new targets for analgesia in burn-related pain.