Robles JTO Supplemental Digital Content 1
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Isyte: Integrated Systems Tool for Eye Gene Discovery
Lens iSyTE: Integrated Systems Tool for Eye Gene Discovery Salil A. Lachke,1,2,3,4 Joshua W. K. Ho,1,4,5 Gregory V. Kryukov,1,4,6 Daniel J. O’Connell,1 Anton Aboukhalil,1,7 Martha L. Bulyk,1,8,9 Peter J. Park,1,5,10 and Richard L. Maas1 PURPOSE. To facilitate the identification of genes associated ther investigation. Extension of this approach to other ocular with cataract and other ocular defects, the authors developed tissue components will facilitate eye disease gene discovery. and validated a computational tool termed iSyTE (integrated (Invest Ophthalmol Vis Sci. 2012;53:1617–1627) DOI: Systems Tool for Eye gene discovery; http://bioinformatics. 10.1167/iovs.11-8839 udel.edu/Research/iSyTE). iSyTE uses a mouse embryonic lens gene expression data set as a bioinformatics filter to select candidate genes from human or mouse genomic regions impli- ven with the advent of high-throughput sequencing, the cated in disease and to prioritize them for further mutational Ediscovery of genes associated with congenital birth defects and functional analyses. such as eye defects remains a challenge. We sought to develop METHODS. Microarray gene expression profiles were obtained a straightforward experimental approach that could facilitate for microdissected embryonic mouse lens at three key devel- the identification of candidate genes for developmental disor- opmental time points in the transition from the embryonic day ders, and, as proof-of-principle, we chose defects involving the (E)10.5 stage of lens placode invagination to E12.5 lens primary ocular lens. Opacification of the lens results in cataract, a leading cause of blindness that affects 77 million persons and fiber cell differentiation. -

A New Mutation in BFSP2 (G1091A) Causes Autosomal Dominant Congenital Lamellar Cataracts
Molecular Vision 2008; 14:1906-1911 <http://www.molvis.org/molvis/v14/a226> © 2008 Molecular Vision Received 17 August 2008 | Accepted 18 October 2008 | Published 24 October 2008 A new mutation in BFSP2 (G1091A) causes autosomal dominant congenital lamellar cataracts Xu Ma,1,2,3 Fei-Feng Li,1,2 Shu-Zhen Wang,4 Chang Gao,1,2 Meng Zhang,2 Si-Quan Zhu4 (The first three authors contributed equally to this work.) 1Graduate School, Peking Union Medical College, Beijing, China; 2Department of Genetics, National Research Institute for Family Planning, Beijing, China; 3WHO Collaborative Center for Research in Human Reproduction, Beijing, China; 4Beijing Tongren Eye Center, Capital Medical University, Beijing, China Purpose: We sought to identify the genetic defect in a four-generation Chinese family with autosomal dominant congenital lamellar cataracts and demonstrate the functional analysis with biosoftware of a candidate gene in the family. Methods: Family history data were recorded. Clinical and ophthalmologic examinations were performed on family members. All the members were genotyped with microsatellite markers at loci considered to be associated with cataracts. Two-point LOD scores were calculated by using the Linkage Software after genotyping. A mutation was detected by using gene-specific primers in direct sequencing. Wild type and mutant proteins were analyzed with Online Bio-Software. Results: Affected members of this family had lamellar cataracts. Linkage analysis was obtained at markers D3S2322 (LOD score [Z]=7.22, recombination fraction [θ]=0.0) and D3S1541 (Z=5.42, θ=0.0). Haplotype analysis indicated that the cataract gene was closely linked to these two markers. -

The Activation of the Glucagon-Like Peptide-1 (GLP-1) Receptor by Peptide and Non-Peptide Ligands
The Activation of the Glucagon-Like Peptide-1 (GLP-1) Receptor by Peptide and Non-Peptide Ligands Clare Louise Wishart Submitted in accordance with the requirements for the degree of Doctor of Philosophy of Science University of Leeds School of Biomedical Sciences Faculty of Biological Sciences September 2013 I Intellectual Property and Publication Statements The candidate confirms that the work submitted is her own and that appropriate credit has been given where reference has been made to the work of others. This copy has been supplied on the understanding that it is copyright material and that no quotation from the thesis may be published without proper acknowledgement. The right of Clare Louise Wishart to be identified as Author of this work has been asserted by her in accordance with the Copyright, Designs and Patents Act 1988. © 2013 The University of Leeds and Clare Louise Wishart. II Acknowledgments Firstly I would like to offer my sincerest thanks and gratitude to my supervisor, Dr. Dan Donnelly, who has been nothing but encouraging and engaging from day one. I have thoroughly enjoyed every moment of working alongside him and learning from his guidance and wisdom. My thanks go to my academic assessor Professor Paul Milner whom I have known for several years, and during my time at the University of Leeds he has offered me invaluable advice and inspiration. Additionally I would like to thank my academic project advisor Dr. Michael Harrison for his friendship, help and advice. I would like to thank Dr. Rosalind Mann and Dr. Elsayed Nasr for welcoming me into the lab as a new PhD student and sharing their experimental techniques with me, these techniques have helped me no end in my time as a research student. -

Core Transcriptional Regulatory Circuitries in Cancer
Oncogene (2020) 39:6633–6646 https://doi.org/10.1038/s41388-020-01459-w REVIEW ARTICLE Core transcriptional regulatory circuitries in cancer 1 1,2,3 1 2 1,4,5 Ye Chen ● Liang Xu ● Ruby Yu-Tong Lin ● Markus Müschen ● H. Phillip Koeffler Received: 14 June 2020 / Revised: 30 August 2020 / Accepted: 4 September 2020 / Published online: 17 September 2020 © The Author(s) 2020. This article is published with open access Abstract Transcription factors (TFs) coordinate the on-and-off states of gene expression typically in a combinatorial fashion. Studies from embryonic stem cells and other cell types have revealed that a clique of self-regulated core TFs control cell identity and cell state. These core TFs form interconnected feed-forward transcriptional loops to establish and reinforce the cell-type- specific gene-expression program; the ensemble of core TFs and their regulatory loops constitutes core transcriptional regulatory circuitry (CRC). Here, we summarize recent progress in computational reconstitution and biologic exploration of CRCs across various human malignancies, and consolidate the strategy and methodology for CRC discovery. We also discuss the genetic basis and therapeutic vulnerability of CRC, and highlight new frontiers and future efforts for the study of CRC in cancer. Knowledge of CRC in cancer is fundamental to understanding cancer-specific transcriptional addiction, and should provide important insight to both pathobiology and therapeutics. 1234567890();,: 1234567890();,: Introduction genes. Till now, one critical goal in biology remains to understand the composition and hierarchy of transcriptional Transcriptional regulation is one of the fundamental mole- regulatory network in each specified cell type/lineage. -

Apc11 (ANAPC11) (NM 001002245) Human Tagged ORF Clone Product Data
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for RC223841L4 Apc11 (ANAPC11) (NM_001002245) Human Tagged ORF Clone Product data: Product Type: Expression Plasmids Product Name: Apc11 (ANAPC11) (NM_001002245) Human Tagged ORF Clone Tag: mGFP Symbol: ANAPC11 Synonyms: APC11; Apc11p; HSPC214 Vector: pLenti-C-mGFP-P2A-Puro (PS100093) E. coli Selection: Chloramphenicol (34 ug/mL) Cell Selection: Puromycin ORF Nucleotide The ORF insert of this clone is exactly the same as(RC223841). Sequence: Restriction Sites: SgfI-MluI Cloning Scheme: ACCN: NM_001002245 ORF Size: 252 bp This product is to be used for laboratory only. Not for diagnostic or therapeutic use. View online » ©2021 OriGene Technologies, Inc., 9620 Medical Center Drive, Ste 200, Rockville, MD 20850, US 1 / 2 Apc11 (ANAPC11) (NM_001002245) Human Tagged ORF Clone – RC223841L4 OTI Disclaimer: Due to the inherent nature of this plasmid, standard methods to replicate additional amounts of DNA in E. coli are highly likely to result in mutations and/or rearrangements. Therefore, OriGene does not guarantee the capability to replicate this plasmid DNA. Additional amounts of DNA can be purchased from OriGene with batch-specific, full-sequence verification at a reduced cost. Please contact our customer care team at [email protected] or by calling 301.340.3188 option 3 for pricing and delivery. The molecular sequence of this clone aligns with the gene accession number as a point of reference only. However, individual transcript sequences of the same gene can differ through naturally occurring variations (e.g. -
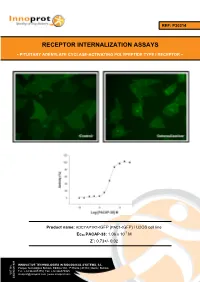
Receptor Internalization Assays
REF: P30214 RECEPTOR INTERNALIZATION ASSAYS - PITUITARY ADENYLATE CYCLASE-ACTIVATING POLYPEPTIDE TYPE I RECEPTOR - Product name: ADCYAP1R1-tGFP (PAC1-tGFP) / U2OS cell line -7 Ec50 PACAP-38: 1.06 x 10 M Z´: 0.73+/- 0.02 INNOVATIVE TECHNOLOGIES IN BIOLOGICAL SYSTEMS, S.L. Parque Tecnológico Bizkaia, Edifício 502, 1ª Planta | 48160 | Derio | Bizkaia Tel.: +34 944005355 | Fax: +34 946579925 VAT No. [email protected] | www.innoprot.com ESB95481909 Product Name: ADCYAP1R1-tGFP_U2OS Reference: P30214 Rep. Official Full Name: Pituitary adenylate cyclase- activating polypeptide type I receptor DNA Accession Number: Gene Bank AY366498 Host Cell: U2OS References: P30214: 2 vials of 3 x 106 proliferative cells P30214-DA: 1 vial of 2 x 106 division-arrested cells Storage: Liquid Nitrogen Assay Briefly description About ADCYAP1R1 Each vial of ADCYAP1R1 Internalization Assay Pituitary adenylate cyclase-activating Cell Line contains U2OS cells stably expressing polypeptide type I receptor, also known as human Pituitary adenylate cyclase-activating PAC1 is a protein that in humans is encoded by polypeptide type I receptor tagged in the N- the ADCYAP1R1 gene. ADCYAP1R1 is a terminus with tGFP protein. membrane-associated protein and shares significant homology with members of the Innoprot’s ADCYAP1R1-tGFP Internalization glucagon/secretin receptor family. This receptor Assay Cell Line has been designed to assay binds pituitary adenylate cyclase activating potential agonists/ antagonists against peptide (PACAP) mediating several biological ADCYAP1R1, modulating its activation and the activities and it is positively coupled to following redistribution process inside the cells. adenylate cyclase. This cell line will allow the image analysis of the stimuli induced by the compounds. -

Early B-Cell Factors Are Required for Specifying Multiple Retinal Cell Types and Subtypes from Postmitotic Precursors
11902 • The Journal of Neuroscience, September 8, 2010 • 30(36):11902–11916 Development/Plasticity/Repair Early B-Cell Factors Are Required for Specifying Multiple Retinal Cell Types and Subtypes from Postmitotic Precursors Kangxin Jin,1,2 Haisong Jiang,1,2 Zeqian Mo,3 and Mengqing Xiang1,2 1Center for Advanced Biotechnology and Medicine and Department of Pediatrics, 2Graduate Program in Molecular Genetics, Microbiology and Immunology, and 3Department of Cell Biology and Neuroscience, University of Medicine and Dentistry of New Jersey-Robert Wood Johnson Medical School, Piscataway, New Jersey 08854 The establishment of functional retinal circuits in the mammalian retina depends critically on the proper generation and assembly of six classes of neurons, five of which consist of two or more subtypes that differ in morphologies, physiological properties, and/or sublaminar positions. How these diverse neuronal types and subtypes arise during retinogenesis still remains largely to be defined at the molecular level. Here we show that all four family members of the early B-cell factor (Ebf) helix-loop-helix transcription factors are similarly expressedduringmouseretinogenesisinseveralneuronaltypesandsubtypesincludingganglion,amacrine,bipolar,andhorizontalcells, and that their expression in ganglion cells depends on the ganglion cell specification factor Brn3b. Misexpressed Ebfs bias retinal precursors toward the fates of non-AII glycinergic amacrine, type 2 OFF-cone bipolar and horizontal cells, whereas a dominant-negative Ebf suppresses the differentiation of these cells as well as ganglion cells. Reducing Ebf1 expression by RNA interference (RNAi) leads to an inhibitory effect similar to that of the dominant-negative Ebf, effectively neutralizes the promotive effect of wild-type Ebf1, but has no impact on the promotive effect of an RNAi-resistant Ebf1. -

Seq2pathway Vignette
seq2pathway Vignette Bin Wang, Xinan Holly Yang, Arjun Kinstlick May 19, 2021 Contents 1 Abstract 1 2 Package Installation 2 3 runseq2pathway 2 4 Two main functions 3 4.1 seq2gene . .3 4.1.1 seq2gene flowchart . .3 4.1.2 runseq2gene inputs/parameters . .5 4.1.3 runseq2gene outputs . .8 4.2 gene2pathway . 10 4.2.1 gene2pathway flowchart . 11 4.2.2 gene2pathway test inputs/parameters . 11 4.2.3 gene2pathway test outputs . 12 5 Examples 13 5.1 ChIP-seq data analysis . 13 5.1.1 Map ChIP-seq enriched peaks to genes using runseq2gene .................... 13 5.1.2 Discover enriched GO terms using gene2pathway_test with gene scores . 15 5.1.3 Discover enriched GO terms using Fisher's Exact test without gene scores . 17 5.1.4 Add description for genes . 20 5.2 RNA-seq data analysis . 20 6 R environment session 23 1 Abstract Seq2pathway is a novel computational tool to analyze functional gene-sets (including signaling pathways) using variable next-generation sequencing data[1]. Integral to this tool are the \seq2gene" and \gene2pathway" components in series that infer a quantitative pathway-level profile for each sample. The seq2gene function assigns phenotype-associated significance of genomic regions to gene-level scores, where the significance could be p-values of SNPs or point mutations, protein-binding affinity, or transcriptional expression level. The seq2gene function has the feasibility to assign non-exon regions to a range of neighboring genes besides the nearest one, thus facilitating the study of functional non-coding elements[2]. Then the gene2pathway summarizes gene-level measurements to pathway-level scores, comparing the quantity of significance for gene members within a pathway with those outside a pathway. -

Dual Proteome-Scale Networks Reveal Cell-Specific Remodeling of the Human Interactome
bioRxiv preprint doi: https://doi.org/10.1101/2020.01.19.905109; this version posted January 19, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Dual Proteome-scale Networks Reveal Cell-specific Remodeling of the Human Interactome Edward L. Huttlin1*, Raphael J. Bruckner1,3, Jose Navarrete-Perea1, Joe R. Cannon1,4, Kurt Baltier1,5, Fana Gebreab1, Melanie P. Gygi1, Alexandra Thornock1, Gabriela Zarraga1,6, Stanley Tam1,7, John Szpyt1, Alexandra Panov1, Hannah Parzen1,8, Sipei Fu1, Arvene Golbazi1, Eila Maenpaa1, Keegan Stricker1, Sanjukta Guha Thakurta1, Ramin Rad1, Joshua Pan2, David P. Nusinow1, Joao A. Paulo1, Devin K. Schweppe1, Laura Pontano Vaites1, J. Wade Harper1*, Steven P. Gygi1*# 1Department of Cell Biology, Harvard Medical School, Boston, MA, 02115, USA. 2Broad Institute, Cambridge, MA, 02142, USA. 3Present address: ICCB-Longwood Screening Facility, Harvard Medical School, Boston, MA, 02115, USA. 4Present address: Merck, West Point, PA, 19486, USA. 5Present address: IQ Proteomics, Cambridge, MA, 02139, USA. 6Present address: Vor Biopharma, Cambridge, MA, 02142, USA. 7Present address: Rubius Therapeutics, Cambridge, MA, 02139, USA. 8Present address: RPS North America, South Kingstown, RI, 02879, USA. *Correspondence: [email protected] (E.L.H.), [email protected] (J.W.H.), [email protected] (S.P.G.) #Lead Contact: [email protected] bioRxiv preprint doi: https://doi.org/10.1101/2020.01.19.905109; this version posted January 19, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. -

PROTEOMIC ANALYSIS of HUMAN URINARY EXOSOMES. Patricia
ABSTRACT Title of Document: PROTEOMIC ANALYSIS OF HUMAN URINARY EXOSOMES. Patricia Amalia Gonzales Mancilla, Ph.D., 2009 Directed By: Associate Professor Nam Sun Wang, Department of Chemical and Biomolecular Engineering Exosomes originate as the internal vesicles of multivesicular bodies (MVBs) in cells. These small vesicles (40-100 nm) have been shown to be secreted by most cell types throughout the body. In the kidney, urinary exosomes are released to the urine by fusion of the outer membrane of the MVBs with the apical plasma membrane of renal tubular epithelia. Exosomes contain apical membrane and cytosolic proteins and can be isolated using differential centrifugation. The analysis of urinary exosomes provides a non- invasive means of acquiring information about the physiological or pathophysiological state of renal cells. The overall objective of this research was to develop methods and knowledge infrastructure for urinary proteomics. We proposed to conduct a proteomic analysis of human urinary exosomes. The first objective was to profile the proteome of human urinary exosomes using liquid chromatography-tandem spectrometry (LC- MS/MS) and specialized software for identification of peptide sequences from fragmentation spectra. We unambiguously identified 1132 proteins. In addition, the phosphoproteome of human urinary exosomes was profiled using the neutral loss scanning acquisition mode of LC-MS/MS. The phosphoproteomic profiling identified 19 phosphorylation sites corresponding to 14 phosphoproteins. The second objective was to analyze urinary exosomes samples isolated from patients with genetic mutations. Polyclonal antibodies were generated to recognize epitopes on the gene products of these genetic mutations, NKCC2 and MRP4. The potential usefulness of urinary exosome analysis was demonstrated using the well-defined renal tubulopathy, Bartter syndrome type I and using the single nucleotide polymorphism in the ABCC4 gene. -

4-6 Weeks Old Female C57BL/6 Mice Obtained from Jackson Labs Were Used for Cell Isolation
Methods Mice: 4-6 weeks old female C57BL/6 mice obtained from Jackson labs were used for cell isolation. Female Foxp3-IRES-GFP reporter mice (1), backcrossed to B6/C57 background for 10 generations, were used for the isolation of naïve CD4 and naïve CD8 cells for the RNAseq experiments. The mice were housed in pathogen-free animal facility in the La Jolla Institute for Allergy and Immunology and were used according to protocols approved by the Institutional Animal Care and use Committee. Preparation of cells: Subsets of thymocytes were isolated by cell sorting as previously described (2), after cell surface staining using CD4 (GK1.5), CD8 (53-6.7), CD3ε (145- 2C11), CD24 (M1/69) (all from Biolegend). DP cells: CD4+CD8 int/hi; CD4 SP cells: CD4CD3 hi, CD24 int/lo; CD8 SP cells: CD8 int/hi CD4 CD3 hi, CD24 int/lo (Fig S2). Peripheral subsets were isolated after pooling spleen and lymph nodes. T cells were enriched by negative isolation using Dynabeads (Dynabeads untouched mouse T cells, 11413D, Invitrogen). After surface staining for CD4 (GK1.5), CD8 (53-6.7), CD62L (MEL-14), CD25 (PC61) and CD44 (IM7), naïve CD4+CD62L hiCD25-CD44lo and naïve CD8+CD62L hiCD25-CD44lo were obtained by sorting (BD FACS Aria). Additionally, for the RNAseq experiments, CD4 and CD8 naïve cells were isolated by sorting T cells from the Foxp3- IRES-GFP mice: CD4+CD62LhiCD25–CD44lo GFP(FOXP3)– and CD8+CD62LhiCD25– CD44lo GFP(FOXP3)– (antibodies were from Biolegend). In some cases, naïve CD4 cells were cultured in vitro under Th1 or Th2 polarizing conditions (3, 4). -

Supplementary Table 1: Adhesion Genes Data Set
Supplementary Table 1: Adhesion genes data set PROBE Entrez Gene ID Celera Gene ID Gene_Symbol Gene_Name 160832 1 hCG201364.3 A1BG alpha-1-B glycoprotein 223658 1 hCG201364.3 A1BG alpha-1-B glycoprotein 212988 102 hCG40040.3 ADAM10 ADAM metallopeptidase domain 10 133411 4185 hCG28232.2 ADAM11 ADAM metallopeptidase domain 11 110695 8038 hCG40937.4 ADAM12 ADAM metallopeptidase domain 12 (meltrin alpha) 195222 8038 hCG40937.4 ADAM12 ADAM metallopeptidase domain 12 (meltrin alpha) 165344 8751 hCG20021.3 ADAM15 ADAM metallopeptidase domain 15 (metargidin) 189065 6868 null ADAM17 ADAM metallopeptidase domain 17 (tumor necrosis factor, alpha, converting enzyme) 108119 8728 hCG15398.4 ADAM19 ADAM metallopeptidase domain 19 (meltrin beta) 117763 8748 hCG20675.3 ADAM20 ADAM metallopeptidase domain 20 126448 8747 hCG1785634.2 ADAM21 ADAM metallopeptidase domain 21 208981 8747 hCG1785634.2|hCG2042897 ADAM21 ADAM metallopeptidase domain 21 180903 53616 hCG17212.4 ADAM22 ADAM metallopeptidase domain 22 177272 8745 hCG1811623.1 ADAM23 ADAM metallopeptidase domain 23 102384 10863 hCG1818505.1 ADAM28 ADAM metallopeptidase domain 28 119968 11086 hCG1786734.2 ADAM29 ADAM metallopeptidase domain 29 205542 11085 hCG1997196.1 ADAM30 ADAM metallopeptidase domain 30 148417 80332 hCG39255.4 ADAM33 ADAM metallopeptidase domain 33 140492 8756 hCG1789002.2 ADAM7 ADAM metallopeptidase domain 7 122603 101 hCG1816947.1 ADAM8 ADAM metallopeptidase domain 8 183965 8754 hCG1996391 ADAM9 ADAM metallopeptidase domain 9 (meltrin gamma) 129974 27299 hCG15447.3 ADAMDEC1 ADAM-like,