Transdermal and Transbuccal Drug Delivery
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Formulation and Evaluation of Transdermal Patch and Gel of Nateglinide
Human Journals Research Article September 2015 Vol.:4, Issue:2 © All rights are reserved by C. Aparna et al. Formulation and Evaluation of Transdermal Patch and Gel of Nateglinide Keywords: Nateglinide, transdermal patch and gel, HPMC, ethyl cellulose, carbopol, PVA, PVP ABSTRACT Anusha Gundeti, C. Aparna*, Dr. Prathima Srinivas The objective of the present work was to formulate Transdermal Drug Delivery systems of Nateglinide, an Department of Pharmaceutics, Sri Venkateshwara antidiabetic drug belonging to meglitinide class with a half life of 1.5 hrs. Transdermal patches containing nateglinide were College of Pharmacy, prepared by solvent casting method using the combinations Affiliated to Osmania University, of HPMC:EC, PVA:PVP, HPMC:Eudragit RS 100, Eudragit RL100:RS100 in different proportions and by incorporating Madhapur, Hyderabad, Telangana -500081, India. different permeation enhancers (polyethylene glycol 400, Su bmission: 7 September 2015 DMSO). The transdermal patches were evaluated for their physicochemical properties like thickness, weight variation, Accepted: 11 September 2015 folding endurance, percentage moisture absorption, percentage Published: 25 September 2015 moisture loss, in-vitro diffusion studies & ex-vivo permeation studies. Transdermal Gel was formulated using HPMC, carbopol 934, carbopol 940 and methyl cellulose. Gels were evaluated for homogeneity, pH, viscosity, drug content, in-vitro diffusion studies & ex-vivo permeation studies. By comparing the drug release F5 (HPMC:EC) formulation was selected as optimized formulation as it could sustain the drug release for 12 hrs i.e. 99.2% when compared to gel. Stability studies were www.ijppr.humanjournals.com carried out according to ICH guidelines and the patches maintained integrity and good physicochemical properties during the study period. -

Mucoadhesive Buccal Drug Delivery System: a Review
REVIEW ARTICLE Am. J. PharmTech Res. 2020; 10(02) ISSN: 2249-3387 Journal home page: http://www.ajptr.com/ Mucoadhesive Buccal Drug Delivery System: A Review Ashish B. Budhrani* , Ajay K. Shadija Datta Meghe College of Pharmacy, Salod (Hirapur), Wardha – 442001, Maharashtra, India. ABSTRACT Current innovation in pharmaceuticals determine the merits of mucoadhesive drug delivery system is particularly relevant than oral control release, for getting local systematic drugs distribution in GIT for a prolong period of time at a predetermined rate. The demerits relative with the oral drug delivery system is the extensive presystemic metabolism, degrade in acidic medium as a result insufficient absorption of the drugs. However parental drug delivery system may beat the downside related with oral drug delivery system but parental drug delivery system has significant expense, least patient compliance and supervision is required. By the buccal drug delivery system the medication are directly pass via into systemic circulation, easy administration without pain, brief enzymatic activity, less hepatic metabolism and excessive bioavailability. This review article is an outline of buccal dosage form, mechanism of mucoadhesion, in-vitro and in-vivo mucoadhesion testing technique. Keywords: Buccal drug delivery system, Mucoadhesive drug delivery system, Mucoadhesion, mucoadhesive polymers, Permeation enhancers, Bioadhesive polymers. *Corresponding Author Email: [email protected] Received 10 February 2020, Accepted 29 February 2020 Please cite this article as: Budhrani AB et al., Mucoadhesive Buccal Drug Delivery System: A Review . American Journal of PharmTech Research 2020. Budhrani et. al., Am. J. PharmTech Res. 2020; 10(02) ISSN: 2249-3387 INTRODUCTION Amongst the numerous routes of drug delivery system, oral drug delivery system is possibly the maximum preferred to the patient. -

Preparation of Mucoadhesive Patches for Buccal Administration of Metoprolol Succinate: in Vitro and in Vivo Drug Release and Bioadhesion
Verma & Chattopadhyay Tropical Journal of Pharmaceutical Research February 2012; 11 (1): 9-17 © Pharmacotherapy Group, Faculty of Pharmacy, University of Benin, Benin City, 300001 Nigeria. All rights reserved . Available online at http://www.tjpr.org http://dx.doi.org/10.4314/tjpr.v11i1.2 Research Article Preparation of Mucoadhesive Patches for Buccal Administration of Metoprolol Succinate: In Vitro and In Vivo Drug Release and Bioadhesion Navneet Verma 1,2* and Pronobesh Chattopadhyay 3 1College of Pharmacy, IFTM University, Moradabad-244001 (U.P.), 2Institute of Pharmacy, Bhagwant University, Ajmer-305004 (Raj.), 3Defence Research Laboratory, Tejpur-784001 (Assam), India Abstract Purpose: To develop mucoadhesive patches for buccal administration of metoprolol succinate and to evaluate their in vitro and in vivo bioadhesion. Methods: The mucoadhesive buccal patches were prepared by solvent casting technique using two different mucoadhesive polymers. The formulations were tested for in vitro drug permeation studies, buccal absorption, in vitro drug release studies, moisture absorption as well as for in vitro and in vivo bioadhesion. Results: The peak detachment force and work of adhesion for MC5 (sodium carboxymethylcellulose, i.e., Na CMC) patch were 0.87 N and 0.451 mJ respectively and the corresponding values for CH5 (chitosan) were 5.15N and 0.987 mJ. Formulation CH5 (prepared with chitosan) showed 67.1 % release, while MC5 (Na CMC) showed drug release of 81.9 % in 6 h. Basic pharmacokinetic parameters such as C max , T max and AUC total varied statistically (p < 0.05) when given by the buccal route compared with that of the solution given by the oral route. -

Buccal Tablets
HIGHLIGHTS OF PRESCRIBING INFORMATION • As a part of the TIRF REMS Access program, fentanyl buccal tablets These highlights do not include all the information needed to use fentanyl may be dispensed only to patients enrolled in the TIRF REMS Access buccal tablets safely and effectively. See full prescribing information for program. For inpatient administration of fentanyl buccal tablets (e.g., fentanyl buccal tablets. hospitals, hospices, and long-term care facilities that prescribe for inpatient use), patient and prescriber enrollment is not required. Fentanyl Buccal Tablets, CII ----------------------DOSAGE AND ADMINISTRATION---------------------- Initial U.S. Approval: 1968 • Patients must require and use around-the-clock opioids when taking WARNING: LIFE-THREATENING RESPIRATORY DEPRESSION; fentanyl buccal tablets. (1) ACCIDENTAL INGESTION; RISKS FROM CYTOCHROME P450 • Use the lowest effective dosage for the shortest duration consistent with 3A4 INTERACTION; RISKS FROM CONCOMITANT USE WITH individual patient treatment goals. (2.1) BENZODIAZEPINES OR OTHER CNS DEPRESSANTS; RISK OF • Individualize dosing based on the severity of pain, patient response, prior MEDICATION ERRORS; ADDICTION, ABUSE, AND MISUSE; analgesic experience, and risk factors for addiction, abuse, and misuse. REMS; and NEONATAL OPIOID WITHDRAWAL SYNDROME (2.1) See full prescribing information for complete boxed warning. • Initial dose of fentanyl buccal tablets: 100 mcg. (2.2) • Serious, life-threatening, and/or fatal respiratory depression has • Initiate titration using multiples of 100 mcg fentanyl buccal tablets. Limit occurred. Monitor closely, especially upon initiation or following a patient access to only one strength of fentanyl buccal tablets at any one dose increase. Due to the risk of fatal respiratory depression, fentanyl time. (2.3) buccal tablets are contraindicated in opioid non-tolerant patients (1) • Individually titrate to a tolerable dose that provides adequate analgesia and in management of acute or postoperative pain, including using single fentanyl buccal tablets. -

Buccal Administration of Estrogens
Europaisches Patentamt European Patent Office GO Publication number: 0 286 581 Office europeen des brevets A1 EUROPEAN PATENT APPLICATION Application number: 88730081.2 mt. a.*: A 61 K 31/565 A 61 K 47/00, A 61 K 9/20 Date of filing: 08.04.88 Priority: 10.04.87 US 37273 @ Applicant: ZETACHRON, INC. Post Office Box 339, 100 North Science Park Road Date of publication of application: State College, PA 16803 (US) 12.10.88 Bulletin 88/41 @ Inventor: Keith, Alec D. Designated Contracting States: 539 Beaumont Drive AT BE CH DE ES FR GB GR IT LI LU NL SE Boalsburg Pennsylvania 16801 (US) Snipes, Wallace C. Deepwood Drive Pine Grove Mills Pennsylvania 16868 (US) @ Representative: UEXKULL & STOLBERG Patentanwalte Beselerstrasse 4 D-2000 Hamburg 52 (DE) @ Buccal administration of estrogens. (g) An adequate blood plasma level of 17-beta-estradiol or ethinyl estradiol is attained in a patient in need of estrogen therapy by administration of a dose of 17-beta-estradiol or ethinyl estradiol or a pharmaceutically acceptable ester thereof not greater than 1 50 micrograms into the vestibule of the buccal cavity of the patient and maintaining the dose in contact with the oral mucosa for a period of time sufficient for transmucosal absorption of a sufficient amount of estrogen to produce a therapeutically effective plasma concentration of estrogen. 00 LO CO 00 CM Q. Ill Bundesdruckerei Berlin 0 286 581 Description BUCCAL ADMINISTRATION OF ESTROGENS BACKGROUND OF THE INVENTION 5 Field of the invention: This invention relates to methods for administering drugs to human patients and more particularly to methods of administering estrogens. -

(10) Patent No.: US 9517190 B2
USO09517190B2 (12) United States Patent (10) Patent No.: US 9,517,190 B2 Johncock et al. (45) Date of Patent: Dec. 13, 2016 (54) COATED TITANIUM DIOXIDE TO REDUCE (2013.01); A61K 9/5021 (2013.01); A61 K THE WHITENING EFFECT ON SKN 9/5089 (2013.01); A61O 1704 (2013.01); C09C I/3661 (2013.01); C09C I/3669 (71) Applicant: Symrise AG, Holzminden (DE) (2013.01); C09C I/3692 (2013.01); A61 K 2800/413 (2013.01); A6 IK 2800/622 (72) Inventors: William Johncock, Reinbek (DE); (2013.01); A61 K 2800/63 (2013.01) Simone Peters, Buchholz (DE): (58) Field of Classification Search Martina Issleib, Hoisdorf (DE); CPC ............................ A61K 9/5021: A61K 9/5089 Jirgen Claus, Bevern (DE) See application file for complete search history. (73) Assignee: Symrise AG, Holzminden (DE) (56) References Cited (*) Notice: Subject to any disclaimer, the term of this patent is extended or adjusted under 35 U.S. PATENT DOCUMENTS U.S.C. 154(b) by 0 days. 3,649,321 A 3, 1972 Durrant et al. (21) Appl. No.: 14/661,4879 8,545,891 B1 10/2013 Halpern et al. (22) Filed: Mar 18, 2015 FOREIGN PATENT DOCUMENTS e JLV9 O O WO 2009/126859 A2 10/2009 (65) Prior Publication Data WO 2012, 110302 A2 8, 2012 US 2015/026551 O A1 Sep. 24, 2015 WO 2012110302 A2 * 8, 2012 ....... A61K 2800,622 * cited by examiner (30) Foreign Application Priority Data Mar. 18, 2014 (EP) ..................................... 14160519 Primary Examiner — Carlos Azpuru (74) Attorney, Agent, or Firm — Dilworth & Barrese, (51) Int. Cl. LLP A6LX 9/50 (2006.01) A6 IK 8/29 (2006.01) A6 IK 8/26 (2006.01) (57) ABSTRACT set, 3,4. -

An Archetype Swing in Transdermal Drug Delivery
Indo American Journal of Pharmaceutical Research, 2017 ISSN NO: 2231-6876 A COMPREHENSIVE REVIEW ON MICRONEEDLES - AN ARCHETYPE SWING IN TRANSDERMAL DRUG DELIVERY G. Ravi*, N. Vishal Gupta, M. P. Gowrav Department of Pharmaceutics, JSS College of Pharmacy, JSS University, Shri Shivarathreeshwara Nagara, Mysuru, Karnataka, India. ARTICLE INFO ABSTRACT Article history Transdermal drug delivery is the non-invasive delivery of medications through the skin Received 23/12/2016 surface into the systemic circulation. The advantage of transdermal drug delivery system is Available online that it is painless technique of administration of drugs. The advantage of transdermal drug 31/01/2017 delivery system is that it is painless technique of administration of drugs. Transdermal drug delivery system can improve the therapeutic efficacy and safety of the drugs because drug Keywords delivered through the skin at a predetermined and controlled rate. Due to the various Microneedles, biomedical benefits, it has attracted many researches. The barrier nature of stratumcorneum Hypodermic Needles, poses a danger to the drug delivery. By using microneedles, a pathway into the human body Transdermal, can be recognized which allow transportation of macromolecular drugs such as insulin or Stratumcorneum, vaccine. These microneedles only penetrate outer layers of the skin, exterior sufficient not to Patch. reach the nerve receptors of the deeper skin. Thus the microneedles supplement is supposed painless and reduces the infection and injuries. Researches from the past few years showed that microneedles have emerged as a novel carrier and considered to be effective for safe and improved delivery of the different drugs. Microneedles development is created a new pathway in the drug delivery field. -

Transdermal Nicotine Maintenance Attenuates the Subjective And
Neuropsychopharmacology (2004) 29, 991–1003 & 2004 Nature Publishing Group All rights reserved 0893-133X/04 $25.00 www.neuropsychopharmacology.org Transdermal Nicotine Maintenance Attenuates the Subjective and Reinforcing Effects of Intravenous Nicotine, but not Cocaine or Caffeine, in Cigarette-Smoking Stimulant Abusers 1 1 ,1,2 Bai-Fang X Sobel , Stacey C Sigmon and Roland R Griffiths* 1Department of Psychiatry and Behavioral Science, Johns Hopkins University School of Medicine, Baltimore, MD, USA; 2Department of Neuroscience, Johns Hopkins University School of Medicine, Baltimore, MD, USA The effects of transdermal nicotine maintenance on the subjective, reinforcing, and cardiovascular effects of intravenously administered cocaine, caffeine, and nicotine were examined using double-blind procedures in nine volunteers with histories of using tobacco, caffeine, and cocaine. Each participant was exposed to two chronic drug maintenance phases (21 mg/day nicotine transdermal patch and placebo transdermal patch). Within each drug phase, the participant received intravenous injections of placebo, cocaine (15 and 30 mg/70 kg), caffeine (200 and 400 mg/70 kg), and nicotine (1.0 and 2.0 mg/70 kg) in mixed order across days. Subjective and cardiovascular data were collected before and repeatedly after drug or placebo injection. Reinforcing effects were also assessed after each injection with a Drug vs Money Multiple-Choice Form. Intravenous cocaine produced robust dose-related increases in subjective and reinforcing effects; these effects were not altered by nicotine maintenance. Intravenous caffeine produced elevations on several subjective ratings; nicotine maintenance did not affect these ratings. Under the placebo maintenance condition, intravenous nicotine produced robust dose-related subjective effects, with maximal increases similar to the high dose of cocaine; nicotine maintenance significantly decreased the subjective and reinforcing effects of intravenous nicotine. -

Buprenorphine / Naloxone Buccal Film
Buprenorphine/Naloxone Buccal Film Monograph Buprenorphine / Naloxone Buccal Film (BUNAVAIL) C-III National PBM Abbreviated Drug Review Sep 2014 VHA Pharmacy Benefits Management Services, Medical Advisory Panel, and VISN Pharmacist Executives The PBM prepares abbreviated reviews to compile information relevant to making formulary decisions. The manufacturer’s labeling should be consulted for detailed drug information. VA clinical experts may provide input on the content. Wider field review is not sought. Documents no longer current will be placed in the Archive section of www.pbm.va.gov. Executive Summary: Buprenorphine / naloxone buccal film (BUNAVAIL, by BioDelivery Sciences) was approved on 6 June 2014 by the Food and Drug Administration (FDA) for the maintenance treatment of opioid dependence, and should be used as part of a complete treatment plan to include counseling and psychosocial support. The main difference between buprenorphine / naloxone buccal film and sublingual tablets is a two-fold greater bioavailability due to greater absorption. Conclusions: Buprenorphine / naloxone buccal film produces buprenorphine bioavailabilities (systemic exposures) similar to those of SUBOXONE (buprenorphine / naloxone) sublingual tablets at approximately half the dose of buprenorphine. The more efficient absorption is achieved by using a trademarked BioErodible MucoAdhesive (BEMA®) technology. The manufacturer claims that the lower buprenorphine doses may “help to reduce the potential for misuse and diversion and potentially lessen the incidence of certain side effects” and that the buccal film may potentially overcome some of the challenges associated with sublingual administration. However, no clinical studies have been performed to support these claims. Potential clinical concerns with the buccal film include dosing and administration confusion when switching between the sublingual formulations and the buccal film, as well as look-alike, sound-alike name confusion with other buprenorphine-containing products. -

A Brief Review on Transdermal Patches
Organic and Medicinal Chemistry International Journal ISSN 2474-7610 Review Article Organic & Medicinal Chem IJ Volume 7 Issue 2 - June 2018 Copyright © All rights are reserved by Nidhi Sharma DOI: 10.19080/OMCIJ.2018.07.555707 A Brief Review on Transdermal Patches Nidhi Sharma* HIMT College of Pharmacy, Greater Noida, India Submission: May 12, 2018; Published: June 05, 2018 *Corresponding author: Nidhi Sharma, HIMT College of Pharmacy, Greater Noida, India, Email: Abstract healingTransdermal to an injured drug area delivery of the system body. was An presentedadvantage toof overcome a transdermal the difficulties drug delivery of drug route delivery over especiallyother types oral of route. delivery A transdermal system such patch as oral, is a topical,medicated i.v., adhesive i.m., etc. ispatch that that the patchis placed provides on the askin controlled to deliver release a specific of the dose medication of medication into the through patient, the usually skin and through into the either bloodstream. a porous It membrane promotes covering a reservoir of medication or through body heat melting thin layers of medication embedded in the adhesive. The main disadvantage to transdermal delivery systems stems from the fact that the skin is a very effective barrier, as a result, only medications whose molecules are small can easily penetrate the skin, so it can be delivered by this method. This review article describes the overall introduction of transdermal patches including type of transdermal patches, method of preparation of transdermal -

Medication Administration
MEDICATION ADMINISTRATION GENERAL CONSIDERATIONS A. Before administering any medication, the EMT should know: 1. What is the medication being used? 2. Does the patient have an allergy to this medication? 3. What is the safe and effective dose? 4. What is the correct administration route? 5. What are the indications? (Why are you using is?) 6. What are the contraindications? (Why or when would you NOT use this medication?) 7. What are the expected effects? 8. What are the adverse effects / side effects? 9. Is the medication expired? B. The “Six Rights” of medication administration: 1. Right patient – is the medication indicated for this patient; no contraindications; no allergies 2. Right drug – the correct name (trade name vs. generic name); correct concentration 3. Right dose 4. Right route 5. Right time – slow IVP vs. rapid IVP 6. Right documentation C. Correct documentation of medications administered and/or IV/IO placement will include: 1. Time of medication administration; IV/IO placement 2. Route of administration 3. Size of catheter (IV/IO) 4. Site location for IV/IO and SQ, IM medication (include unsuccessful IV/IO attempt locations) 5. Dose or volume infused 6. Time of infusion as indicated (e.g., rapid IVP, infused over 10 minutes, etc.) 7. Name of EMT responsible 8. Any complications and steps made to correct 9. Patient’s response to treatment D. Use of a medication simply because it is in the protocol is not an acceptable standard of medical care. When there are questions about medication administration, consult medical control. ORAL ADMINSTRATION To administer an oral (PO) medication ensure that the patient has an intact gag reflex and place the patient in a seated or semi-seated position. -
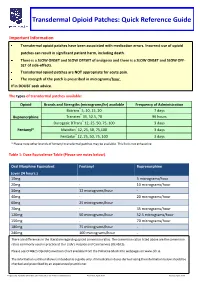
Transdermal Opioid Patches: Quick Reference Guide
Transdermal Opioid Patches: Quick Reference Guide Important Information Transdermal opioid patches have been associated with medication errors. Incorrect use of opioid patches can result in significant patient harm, including death. There is a SLOW ONSET and SLOW OFFSET of analgesia and there is a SLOW ONSET and SLOW OFF- SET of side-effects. Transdermal opioid patches are NOT appropriate for acute pain. The strength of the patch is prescribed in micrograms/hour. If in DOUBT seek advice. The types of transdermal patches available: Opioid Brands and Strengths (micrograms/hr) available Frequency of Administration Butrans® 5, 10, 15, 20 7 days Buprenorphine Transtec® 35, 52.5, 70 96 hours Durogesic DTrans® 12, 25, 50, 75, 100 3 days Fentanyl* Matrifen® 12, 25, 50, 75,100 3 days Fentadur® 12, 25, 50, 75, 100 3 days * Please note other brands of fentanyl transdermal patches may be available. This list is not exhaustive. Table 1: Dose Equivalence Table (Please see notes below). Oral Morphine Equivalent Fentanyl Buprenorphine (over 24 hours.) 10mg 5 micrograms/hour 20mg - 10 micrograms/hour 30mg 12 micrograms/hour - 40mg - 20 micrograms/hour 60mg 25 micrograms/hour 70mg - 35 micrograms/hour 120mg 50 micrograms/hour 52.5 micrograms/hour 150mg - 70 micrograms/hour 180mg 75 micrograms/hour - 240mg 100 micrograms/hour - There are differences in the literature regarding opioid conversion ratios. The conversion ratios listed above are the conversion ratios commonly used in practice at Our Lady’s Hospice and Care Service (OLH&CS). Please see OLH&CS Opioid Conversion Chart available from the Palliative Meds Info webpages on www.olh.ie.