Catalytic Asymmetric Ketone and Alkene Reductions Using Transition Metal Complexes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
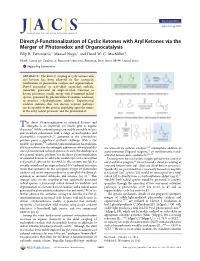
Direct Β‑Functionalization of Cyclic Ketones with Aryl Ketones Via the Merger of Photoredox and Organocatalysis Filip R
Communication pubs.acs.org/JACS Direct β‑Functionalization of Cyclic Ketones with Aryl Ketones via the Merger of Photoredox and Organocatalysis Filip R. Petronijevic,́† Manuel Nappi,† and David W. C. MacMillan* Merck Center for Catalysis at Princeton University, Princeton, New Jersey 08544, United States *S Supporting Information ABSTRACT: The direct β-coupling of cyclic ketones with aryl ketones has been achieved via the synergistic combination of photoredox catalysis and organocatalysis. Diaryl oxymethyl or aryl−alkyl oxymethyl radicals, transiently generated via single-electron reduction of ketone precursors, readily merge with β-enaminyl radical species, generated by photon-induced enamine oxidation, to produce γ-hydroxyketone adducts. Experimental evidence indicates that two discrete reaction pathways can be operable in this process depending upon the nature of the ketyl radical precursor and the photocatalyst. he direct β-functionalization of saturated ketones and T aldehydes is an important yet elusive goal in organic chemistry.1 While carbonyl groups are readily amenable to ipso- and α-carbon substitution with a range of nucleophiles and electrophiles respectively,2,3 activation at the β-methylene position poses a significant synthetic challenge. With a few notable exceptions,1,4 carbonyl β-functionalization has tradition- ally been restricted to the conjugate addition of soft nucleophiles are accessed via carbene catalysis,9,10 nucleophilic addition of into α,β-unsaturated carbonyl systems. As such, the development acetal-protected Grignard reagents,11 or stoichiometric metal- 5 − of a general catalytic platform for the direct β-functionalization activated homoenolate equivalents.12 16 of saturated ketones or aldehydes would represent a conceptual Drawing from the mechanistic insights gained in the course of and practical advance for the field. -

Aldehydes and Ketones
12 Aldehydes and Ketones Ethanol from alcoholic beverages is first metabolized to acetaldehyde before being broken down further in the body. The reactivity of the carbonyl group of acetaldehyde allows it to bind to proteins in the body, the products of which lead to tissue damage and organ disease. Inset: A model of acetaldehyde. (Novastock/ Stock Connection/Glow Images) KEY QUESTIONS 12.1 What Are Aldehydes and Ketones? 12.8 What Is Keto–Enol Tautomerism? 12.2 How Are Aldehydes and Ketones Named? 12.9 How Are Aldehydes and Ketones Oxidized? 12.3 What Are the Physical Properties of Aldehydes 12.10 How Are Aldehydes and Ketones Reduced? and Ketones? 12.4 What Is the Most Common Reaction Theme of HOW TO Aldehydes and Ketones? 12.1 How to Predict the Product of a Grignard Reaction 12.5 What Are Grignard Reagents, and How Do They 12.2 How to Determine the Reactants Used to React with Aldehydes and Ketones? Synthesize a Hemiacetal or Acetal 12.6 What Are Hemiacetals and Acetals? 12.7 How Do Aldehydes and Ketones React with CHEMICAL CONNECTIONS Ammonia and Amines? 12A A Green Synthesis of Adipic Acid IN THIS AND several of the following chapters, we study the physical and chemical properties of compounds containing the carbonyl group, C O. Because this group is the functional group of aldehydes, ketones, and carboxylic acids and their derivatives, it is one of the most important functional groups in organic chemistry and in the chemistry of biological systems. The chemical properties of the carbonyl group are straightforward, and an understanding of its characteristic reaction themes leads very quickly to an understanding of a wide variety of organic reactions. -

Aldehydes and Ketone
ALDEHYDES AND KETONE www.gneet.com ALDEHYDES AND KETONES In aldehydes, the carbonyl group is linked to either two hydrogen atom or one hydrogen atom and one carbon containing group such as alkyl, aryl or aralkyl group Examples * In ketones, the carbonyl group is linked to two carbon containing groups which may be same or different alkyl, aryl group. If two R and R’ groups are same, the ketone is called simple or symmetrical ketone and if R and R’ are different, then ketone is known as mixed or an unsymmetrical ketone. STRUCTURE Carbonyl carbon of both aldehyde and ketones is sp2 – hybridised, One of the three sp2 hybridised orbital get involved in σ- bond formation with half –filled p-orbital of oxygen atom whereas rest of the two are consumed in σ-bond formation with hydrogen and carbon depending on the structure of aldehyde or ketone. Unhybridised p-orbital of carbonyl carbon form π-bond with another half-filled p-orbital of oxygen atom by sideways overlapping. ISOMERISM IN ALDEHYDES AND KETONES (a) Chain isomerism: Aldehydes ( with 4 or more carbon atoms) and ketone ( with 5 or more carbon atoms) show chain isomerism. Example i) C4H8O CH3-CH2-CH2-CHO ( butanal) 1 ALDEHYDES AND KETONE www.gneet.com ii) C5H10O (b) Position isomerism: aliphatic aldehydes do not show position isomerism, because –CHO group is always present at the end of carbon chain. Aromatic aldehyde show position isomerism. Example (c) Metamerism: Higher ketones show metamerism due to presence of different alkyl groups attached to the same functional group C5H10O (d) Functional isomerism : Aldehydes and ketones show functional isomerism in them. -

Chapter 14 – Aldehydes and Ketones
Chapter 14 – Aldehydes and Ketones 14.1 Structures and Physical Properties of Aldehydes and Ketones Ketones and aldehydes are related in that they each possess a C=O (carbonyl) group. They differ in that the carbonyl carbon in ketones is bound to two carbon atoms (RCOR’), while that in aldehydes is bound to at least one hydrogen (H2CO and RCHO). Thus aldehydes always place the carbonyl group on a terminal (end) carbon, while the carbonyl group in ketones is always internal. Some common examples include (common name in parentheses): O O H HH methanal (formaldehyde) trans-3-phenyl-2-propenal (cinnamaldehyde) preservative oil of cinnamon O O propanone (acetone) 3-methylcyclopentadecanone (muscone) nail polish remover a component of one type of musk oil Simple aldehydes (e.g. formaldehyde) typically have an unpleasant, irritating odor. Aldehydes adjacent to a string of double bonds (e.g. 3-phenyl-2-propenal) frequently have pleasant odors. Other examples include the primary flavoring agents in oil of bitter almond (Ph- CHO) and vanilla (C6H3(OH)(OCH3)(CHO)). As your book says, simple ketones have distinctive odors (similar to acetone) that are typically not unpleasant in low doses. Like aldehydes, placing a collection of double bonds adjacent to a ketone carbonyl generally makes the substance more fragrant. The primary flavoring agent in oil of caraway is just a such a ketone. 2 O oil of carraway Because the C=O group is polar, small aldehydes and ketones enjoy significant water solubility. They are also quite soluble in typical organic solvents. 14.2 Naming Aldehydes and Ketones Aldehydes The IUPAC names for aldehydes are obtained by using rules similar to those we’ve seen for other functional groups (e.g. -

Reactions of Aromatic Compounds Just Like an Alkene, Benzene Has Clouds of Electrons Above and Below Its Sigma Bond Framework
Reactions of Aromatic Compounds Just like an alkene, benzene has clouds of electrons above and below its sigma bond framework. Although the electrons are in a stable aromatic system, they are still available for reaction with strong electrophiles. This generates a carbocation which is resonance stabilized (but not aromatic). This cation is called a sigma complex because the electrophile is joined to the benzene ring through a new sigma bond. The sigma complex (also called an arenium ion) is not aromatic since it contains an sp3 carbon (which disrupts the required loop of p orbitals). Ch17 Reactions of Aromatic Compounds (landscape).docx Page1 The loss of aromaticity required to form the sigma complex explains the highly endothermic nature of the first step. (That is why we require strong electrophiles for reaction). The sigma complex wishes to regain its aromaticity, and it may do so by either a reversal of the first step (i.e. regenerate the starting material) or by loss of the proton on the sp3 carbon (leading to a substitution product). When a reaction proceeds this way, it is electrophilic aromatic substitution. There are a wide variety of electrophiles that can be introduced into a benzene ring in this way, and so electrophilic aromatic substitution is a very important method for the synthesis of substituted aromatic compounds. Ch17 Reactions of Aromatic Compounds (landscape).docx Page2 Bromination of Benzene Bromination follows the same general mechanism for the electrophilic aromatic substitution (EAS). Bromine itself is not electrophilic enough to react with benzene. But the addition of a strong Lewis acid (electron pair acceptor), such as FeBr3, catalyses the reaction, and leads to the substitution product. -

20 More About Oxidation–Reduction Reactions
More About 20 Oxidation–Reduction Reactions OOC n important group of organic reactions consists of those that O A involve the transfer of electrons C from one molecule to another. Organic chemists H OH use these reactions—called oxidation–reduction reactions or redox reactions—to synthesize a large O variety of compounds. Redox reactions are also important C in biological systems because many of these reactions produce HH energy. You have seen a number of oxidation and reduction reactions in other chapters, but discussing them as a group will give you the opportunity to CH3OH compare them. In an oxidation–reduction reaction, one compound loses electrons and one com- pound gains electrons. The compound that loses electrons is oxidized, and the one that gains electrons is reduced. One way to remember the difference between oxidation and reduction is with the phrase “LEO the lion says GER”: Loss of Electrons is Oxi- dation; Gain of Electrons is Reduction. The following is an example of an oxidation–reduction reaction involving inorganic reagents: Cu+ + Fe3+ ¡ Cu2+ + Fe2+ In this reaction,Cu+ loses an electron, so Cu+ is oxidized. Fe3+ gains an electron, so Fe3+ is reduced. The reaction demonstrates two important points about oxidation– reduction reactions. First, oxidation is always coupled with reduction. In other words, a compound cannot gain electrons (be reduced) unless another compound in the reaction simultaneously loses electrons (is oxidized). Second, the compound that is oxidized (Cu+) is called the reducing agent because it loses the electrons that are used to reduce the other compound (Fe3+). Similarly, the compound that is reduced (Fe3+) is called the oxidizing agent because it gains the electrons given up by the other compound (Cu+) when it is oxidized. -

Effects of Ketone Bodies on Brain Metabolism and Function In
International Journal of Molecular Sciences Review Effects of Ketone Bodies on Brain Metabolism and Function in Neurodegenerative Diseases Nicole Jacqueline Jensen 1,* , Helena Zander Wodschow 1, Malin Nilsson 1 and Jørgen Rungby 1,2 1 Department of Endocrinology, Bispebjerg University Hospital, 2400 Copenhagen, Denmark; [email protected] (H.Z.W.); malin.sofi[email protected] (M.N.); [email protected] (J.R.) 2 Copenhagen Center for Translational Research, Copenhagen University Hospital, Bispebjerg and Frederiksberg, 2400 Copenhagen, Denmark * Correspondence: [email protected] Received: 4 November 2020; Accepted: 18 November 2020; Published: 20 November 2020 Abstract: Under normal physiological conditions the brain primarily utilizes glucose for ATP generation. However, in situations where glucose is sparse, e.g., during prolonged fasting, ketone bodies become an important energy source for the brain. The brain’s utilization of ketones seems to depend mainly on the concentration in the blood, thus many dietary approaches such as ketogenic diets, ingestion of ketogenic medium-chain fatty acids or exogenous ketones, facilitate significant changes in the brain’s metabolism. Therefore, these approaches may ameliorate the energy crisis in neurodegenerative diseases, which are characterized by a deterioration of the brain’s glucose metabolism, providing a therapeutic advantage in these diseases. Most clinical studies examining the neuroprotective role of ketone bodies have been conducted in patients with Alzheimer’s disease, where brain imaging studies support the notion of enhancing brain energy metabolism with ketones. Likewise, a few studies show modest functional improvements in patients with Parkinson’s disease and cognitive benefits in patients with—or at risk of—Alzheimer’s disease after ketogenic interventions. -

Plasma 'Ketone' Levelin the Diagnosis and Treatment of Diabetic Acidosis
Postgrad. med. J. (October 1968) 44, 799-802. Postgrad Med J: first published as 10.1136/pgmj.44.516.799 on 1 October 1968. Downloaded from Plasma 'ketone' level in the diagnosis and treatment of diabetic acidosis M. S. KNAPP MARGARET E. HORN M.D., M.R.C.P. M.B., M.R.C.P. Lecturer in Medicine, University ofBristol Medical Registrar, Bristol Royal Hospital All patients in possible diabetic acidosis admit- Summary ted to one of the medical firms at the Bristol A simple method of estimating the plasma Royal Infirmary over a 3-year period had an 'ketone' level with 'Acetest' tablets has been used estimation of the plasma 'ketone' level performed in the diagnosis and treatment of diabetic acid- by the clinical staff in the ward side-room. In osis. The method is valuable when used in those patients treated for a moderate or severe association with full biochemical facilities, but it diabetic keto-acidosis the estimation was is especially useful in situations where these can- repeated, usually every four hours. The second not be easily obtained. and subsequent doses of insulin were based to a considerable extent on the plasma 'ketone' titre. Introduction If the titre had not fallen or had risen, an in- In a patient suspected of having diabetic keto- creased dose of insulin was given. A fall in titre acidosis it is useful to be able to rapidly confirm indicated a satisfactory response and either a Protected by copyright. the diagnosis. In this situation we have used a similar dose or a smaller one was used, depending simple side-room method for the detection of on previous blood sugar results. -

Chapter 19 the Chemistry of Aldehydes and Ketones. Addition Reactions
Instructor Supplemental Solutions to Problems © 2010 Roberts and Company Publishers Chapter 19 The Chemistry of Aldehydes and Ketones. Addition Reactions Solutions to In-Text Problems 19.1 (b) (d) (e) (g) 19.2 (a) 2-Propanone (d) (E)-3-Ethoxy-2-propenal (f) 4,4-Dimethyl-2,5-cyclohexadienone 19.3 (b) 2-Cyclohexenone has a lower carbonyl stretching frequency because its two double bonds are conjugated. 19.4 (b) The compound is 2-butanone: (c) The high frequency of the carbonyl absorption suggests a strained ring. (See Eq. 19.4, text p. 897.) In fact, cyclobutanone matches the IR stretching frequency perfectly and the NMR fits as well: 19.6 The structure and CMR assignments of 2-ethylbutanal are shown below. The two methyl groups are chemically equivalent, and the two methylene groups are chemically equivalent; all carbons with different CMR chemical shifts are chemically nonequivalent. INSTRUCTOR SUPPLEMENTAL SOLUTIONS TO PROBLEMS • CHAPTER 19 2 19.7 (a) The double bonds in 2-cyclohexenone are conjugated, but the double bonds in 3-cyclohexenone are not. Consequently, 2-cyclohexenone has the UV spectrum with the greater lmax. 19.9 Compound A, vanillin, should have a p T p* absorption at a greater lmax when dissolved in NaOH solution because the resulting phenolate can delocalize into the carboxaldehyde group; the resulting phenolate from compound B, isovanillin, on the other hand, can only delocalize in the aromatic ring. 19.11 The mass spectrum of 2-heptanone should have major peaks at m/z = 43 (from a-cleavage), 71 (from inductive cleavage), and 58 (from McLafferty rearrangement). -

Hydration of Ketones and Aldehydes
Hydration of Ketones and Aldehydes H H H O O H H H H HO OH H2O O O HO OH O a hydrated ketone H H O HO OH H2O Keq = 2000 uncrowded H H H H Aldehydes exist as partial hydrates in aqueous solution: O HO OH H2O Keq = 1 more crowded Me H Me H O H O HO OH Ketones generally do not favor hydration: 2 Keq = 0.002 very crowded Me Me Me Me Nucleophilic Addition of "O–" to Carbonyl Groups Na O O OH H+ (workup) O HO Na + H2O Me Me Me Me Me Me OH OH Me Me A hydrated ketone Starting material! pKa of H2O: approx. 16 pKa of alkoxide: 16-20 Na O O OH H+ (workup) O MeO Na + MeOH Me Me Me Me Me Me OMe OMe Me Me A hemiacetal Starting material! (usually unstable) pKa of MeOH: approx. 15 pKa of alkoxide: 16-20 Hydration of Ketones and Aldehydes 18 O 18 O H2 O + Me Me H cat. Me Me H H H 18O 18O H H H Me Me H H Me 18 18 O O H O OH O H H Me 18O Me Me H H H18O OH H Me Me 18 H O a hydrated ketone Me Me H 18 H O OH2 18 O 18O Me Me Me Me Addition of Alcohols to Carbonyl Groups: Acetal Formation O ROH, H+ RO OR Me Me Me Me An acetal MeOH H+ Me Me H H Me O O MeO OH H+ Me MeOHMe Me MeO OH MeOH Me Me a hemiacetal MeO OMe H Me Me Me MeO OH2 Me Me MeO OMe O Me Me Me An acetal Me MeOH H2O Acetals as Carbonyl Protecting Groups O OH MeMgCl Me Me Me Me Me MeO OMe MeMgCl No reaction Me Me O Br O Li–Bu O P(Ph)3 Br (Ph)3P X (Ph)3P Me Me Me Butyllithium will react with the ketone, and the reagent will react with itself! O MeO OMe MeOH, H+ MeO OMe (Ph) P Br Br 3 Me Me Me A stable reagent O O MeO OMe + H2O, H Me Me Cyclic Hemiacetals MeOH H+ Me H Me MeOH Me H MeO OH O O MeO OH Me -

IR Handout.Pdf
Infrared spectra: It is important to remember that the absence of an absorption band can often provide more information about the structure of a compound than the presence of a band. Be careful to avoid focusing on selected absorption bands and overlooking others. Use the examples linked to the table to see the profile and intensity of bands. Look for absorption bands in decreasing order of importance: 1.the C-H absorption(s) between 3100 and 2850 cm-1. An absorption above 3000 cm-1 indicates C=C, either alkene or aromatic. Confirm the aromatic ring by finding peaks at 1600 and 1500 cm-1 and C-H out-of-plane bending to give substitution patterns below 900 cm-1. Confirm alkenes with an absorption generally at 1640-1680 cm-1. C-H absorption between 3000 and 2850 cm-1 is due to aliphatic hydrogens. 2. the carbonyl (C=O) absorption between 1690-1760cm-1; this strong band indicates either an aldehyde, ketone, carboxylic acid, ester, amide, anhydride or acyl halide. The an aldehyde may be confirmed with C-H absorption from 2840 to 2720 cm-1. 3. the O-H or N-H absorption between 3200 and 3600 cm-1. This indicates either an alcohol, N-H containing amine or amide, or carboxylic acid. For -NH2 a doublet will be observed. 4.the C-O absorption between 1080 and 1300 cm-1. These peaks are normally rounded like the O-H and N-H peak in 3. and are prominent. Carboxylic acids, esters, ethers, alcohols and anhydrides all containing this peak. -

Chapter 14 Aldehydes, Ketones, and Chiral Molecules If a Carbonyl Group Is Attached to at Least One H Atom, It Is Called an Aldehyde
Chapter 14 Aldehydes, Ketones, and Chiral Molecules If a carbonyl group is attached to at least one H atom, it is called an aldehyde If a carbonyl group is attached to two carbon atoms it is called ketone An aldehyde is named dropping the e in the alkane and adding al Ketones are named by replacing the e in the alkane name with one; the location of the carbonyl carbon is indicated by a number 2- B O A . cyclohexanone . 2-pentanone Several naturally occurring aldehydes and ketones are used as flavorings for foods and as fragrances. O C H benzaldehyde: In acid it is cherry flavor; otherwise it is used as almond flavor; used in amaretto O 4-hydroxy-3-methoxybenzaldehyde CH=CH C H 2,3- butanedione; butter flavor nail polish remover Draw the structural formulas for each of the following: A. 4-Methylpentanal B. 2,3-Dichloropropanal C. 3-Methyl-2-butanone Classify each as an 1) aldehyde, 2) ketone, 3) alcohol, or 4) ether. A. B. CH3─O─CH3 C. D . Physical Properties of Aldehydes and Ketones The carbonyl group is polar However it does not have H on the oxygen atom and cannot form hydrogen bonds Aldehydes and ketones have attractions between polar groups and consequently have higher boiling points than alkanes and ethers of similar size but lower boiling points than alcohols of similar size because they can’t form hydrogen bonds increasing size Reactivity of Aldehydes and Ketones Aldehydes are far more reactive than ketones. They are easily oxidized by the oxygen in the air to carboxylic acids and in water or in alcohols they often hydrate or add