Carboxylic Acids a Carbonyl with One OH Attached Is Called a Carboxylic
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
The Degradation of Carboxylic Acid Salts by Means of Halogen the Hunsdieceerreaction Robert G
THE DEGRADATION OF CARBOXYLIC ACID SALTS BY MEANS OF HALOGEN THE HUNSDIECEERREACTION ROBERT G. JOHNSON Department of Chemistry, Xavier University, Cincinnati 7, Ohio AND ROBERT K. INGHAM Department of Chemistry, Ohio University, Athens, Ohio Received November 17, 1966 CONTENTS I. Introduction.. ...... 11. Synthetic applicatio A. Aliphatic halides B. Alicyclic halides. C. Aromatic halides D. Heterocyclic hali 111. Materials and methods ....................................... 248 IV. Mechanisms., ...... V. Related reactions. ....... ......................................... 259 VI. Summary.. ......... VII. References.. ................................................................. 263 VIII. Addendum. ........................... .. 268 I. INTRODUCTION The degradation of a carboxylic acid salt in anhydrous medium by means of halogen to a halide of one less carbon atom than the original acid can be ex- pressed by the equation: RCOOM + Xz -+ RX + COz + MX Largely because of the extensive contributions of the Hunsdieckers (96, 97, 98, 99, 100, 101, 102) to our knowledge of this reaction, many chemists refer to the reaction under discussion as the “Hunsdiecker reaction.” Others designate the reaction by the names “silver salt reaction” or “silver salt-halogen reac- tion.” It has been proposed (95) that the name “Borodine reaction” be used, in recognition of the discovery of the reaction by Borodine (33). The authors of a recent book (179) incorrectly name this reaction M the Simonini reaction. The reviewers favor the use of “Hunsdiecker reaction,” if a name is to be given to this reaction, because of the many instances of the use of this termi- nology already in the literature and because of the large amount of developmental work carried out by the Hunsdieckers. The often-used descriptive names are contraindicated by the fact that the reaction is successful with salts other than silver salts, halogens other than bromine can be used, and the reaction of salts 219 220 ROBERT G. -
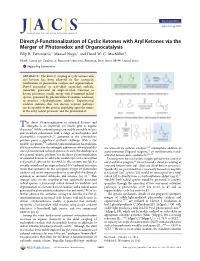
Direct Β‑Functionalization of Cyclic Ketones with Aryl Ketones Via the Merger of Photoredox and Organocatalysis Filip R
Communication pubs.acs.org/JACS Direct β‑Functionalization of Cyclic Ketones with Aryl Ketones via the Merger of Photoredox and Organocatalysis Filip R. Petronijevic,́† Manuel Nappi,† and David W. C. MacMillan* Merck Center for Catalysis at Princeton University, Princeton, New Jersey 08544, United States *S Supporting Information ABSTRACT: The direct β-coupling of cyclic ketones with aryl ketones has been achieved via the synergistic combination of photoredox catalysis and organocatalysis. Diaryl oxymethyl or aryl−alkyl oxymethyl radicals, transiently generated via single-electron reduction of ketone precursors, readily merge with β-enaminyl radical species, generated by photon-induced enamine oxidation, to produce γ-hydroxyketone adducts. Experimental evidence indicates that two discrete reaction pathways can be operable in this process depending upon the nature of the ketyl radical precursor and the photocatalyst. he direct β-functionalization of saturated ketones and T aldehydes is an important yet elusive goal in organic chemistry.1 While carbonyl groups are readily amenable to ipso- and α-carbon substitution with a range of nucleophiles and electrophiles respectively,2,3 activation at the β-methylene position poses a significant synthetic challenge. With a few notable exceptions,1,4 carbonyl β-functionalization has tradition- ally been restricted to the conjugate addition of soft nucleophiles are accessed via carbene catalysis,9,10 nucleophilic addition of into α,β-unsaturated carbonyl systems. As such, the development acetal-protected Grignard reagents,11 or stoichiometric metal- 5 − of a general catalytic platform for the direct β-functionalization activated homoenolate equivalents.12 16 of saturated ketones or aldehydes would represent a conceptual Drawing from the mechanistic insights gained in the course of and practical advance for the field. -

Aldehydes and Ketones
12 Aldehydes and Ketones Ethanol from alcoholic beverages is first metabolized to acetaldehyde before being broken down further in the body. The reactivity of the carbonyl group of acetaldehyde allows it to bind to proteins in the body, the products of which lead to tissue damage and organ disease. Inset: A model of acetaldehyde. (Novastock/ Stock Connection/Glow Images) KEY QUESTIONS 12.1 What Are Aldehydes and Ketones? 12.8 What Is Keto–Enol Tautomerism? 12.2 How Are Aldehydes and Ketones Named? 12.9 How Are Aldehydes and Ketones Oxidized? 12.3 What Are the Physical Properties of Aldehydes 12.10 How Are Aldehydes and Ketones Reduced? and Ketones? 12.4 What Is the Most Common Reaction Theme of HOW TO Aldehydes and Ketones? 12.1 How to Predict the Product of a Grignard Reaction 12.5 What Are Grignard Reagents, and How Do They 12.2 How to Determine the Reactants Used to React with Aldehydes and Ketones? Synthesize a Hemiacetal or Acetal 12.6 What Are Hemiacetals and Acetals? 12.7 How Do Aldehydes and Ketones React with CHEMICAL CONNECTIONS Ammonia and Amines? 12A A Green Synthesis of Adipic Acid IN THIS AND several of the following chapters, we study the physical and chemical properties of compounds containing the carbonyl group, C O. Because this group is the functional group of aldehydes, ketones, and carboxylic acids and their derivatives, it is one of the most important functional groups in organic chemistry and in the chemistry of biological systems. The chemical properties of the carbonyl group are straightforward, and an understanding of its characteristic reaction themes leads very quickly to an understanding of a wide variety of organic reactions. -

The Relative Rates of Thiol–Thioester Exchange and Hydrolysis for Alkyl and Aryl Thioalkanoates in Water
Orig Life Evol Biosph (2011) 41:399–412 DOI 10.1007/s11084-011-9243-4 PREBIOTIC CHEMISTRY The Relative Rates of Thiol–Thioester Exchange and Hydrolysis for Alkyl and Aryl Thioalkanoates in Water Paul J. Bracher & Phillip W. Snyder & Brooks R. Bohall & George M. Whitesides Received: 14 April 2011 /Accepted: 16 June 2011 / Published online: 5 July 2011 # Springer Science+Business Media B.V. 2011 Abstract This article reports rate constants for thiol–thioester exchange (kex), and for acid- mediated (ka), base-mediated (kb), and pH-independent (kw) hydrolysis of S-methyl thioacetate and S-phenyl 5-dimethylamino-5-oxo-thiopentanoate—model alkyl and aryl thioalkanoates, respectively—in water. Reactions such as thiol–thioester exchange or aminolysis could have generated molecular complexity on early Earth, but for thioesters to have played important roles in the origin of life, constructive reactions would have needed to compete effectively with hydrolysis under prebiotic conditions. Knowledge of the kinetics of competition between exchange and hydrolysis is also useful in the optimization of systems where exchange is used in applications such as self-assembly or reversible binding. For the alkyl thioester S-methyl thioacetate, which has been synthesized in −5 −1 −1 −1 −1 −1 simulated prebiotic hydrothermal vents, ka = 1.5×10 M s , kb = 1.6×10 M s , and −8 −1 kw = 3.6×10 s . At pH 7 and 23°C, the half-life for hydrolysis is 155 days. The second- order rate constant for thiol–thioester exchange between S-methyl thioacetate and 2- −1 −1 sulfonatoethanethiolate is kex = 1.7 M s . -

Chapter 7, Haloalkanes, Properties and Substitution Reactions of Haloalkanes Table of Contents 1
Chapter 7, Haloalkanes, Properties and Substitution Reactions of Haloalkanes Table of Contents 1. Alkyl Halides (Haloalkane) 2. Nucleophilic Substitution Reactions (SNX, X=1 or 2) 3. Nucleophiles (Acid-Base Chemistry, pka) 4. Leaving Groups (Acid-Base Chemistry, pka) 5. Kinetics of a Nucleophilic Substitution Reaction: An SN2 Reaction 6. A Mechanism for the SN2 Reaction 7. The Stereochemistry of SN2 Reactions 8. A Mechanism for the SN1 Reaction 9. Carbocations, SN1, E1 10. Stereochemistry of SN1 Reactions 11. Factors Affecting the Rates of SN1 and SN2 Reactions 12. --Eliminations, E1 and E2 13. E2 and E1 mechanisms and product predictions In this chapter we will consider: What groups can be replaced (i.e., substituted) or eliminated The various mechanisms by which such processes occur The conditions that can promote such reactions Alkyl Halides (Haloalkane) An alkyl halide has a halogen atom bonded to an sp3-hybridized (tetrahedral) carbon atom The carbon–chlorine and carbon– bromine bonds are polarized because the halogen is more electronegative than carbon The carbon-iodine bond do not have a per- manent dipole, the bond is easily polarizable Iodine is a good leaving group due to its polarizability, i.e. its ability to stabilize a charge due to its large atomic size Generally, a carbon-halogen bond is polar with a partial positive () charge on the carbon and partial negative () charge on the halogen C X X = Cl, Br, I Different Types of Organic Halides Alkyl halides (haloalkanes) sp3-hybridized Attached to Attached to Attached -

Acid-Base Interactions in Wetting
J. rldlresiortSci. Tetltttol. \irl. -5.No. l. pp.57-69 ( l99l ) o VsP t9el. Acid-baseinteractions in wetting GEORGE M. WHITESIDES,*HANS A. BIEBUYCK.JOHN P.FOLKERS andKEVIN L. PRIME Departntent of Chentistry,Harvard Universiry',Cambridge, hlA 02138,USA Revisedversion received 27 Aueust1990 Abstract-The studv of the ionizationof carboxylicacid groups at the interfacebetween organic solidsand water demonstratesbroad similaritiesto the ionizationsof thesegroups in homogeneous aqueoussolution, but with importantsystematic differences. Creation of a chargedgroup from a neutralone by protonationor deprotonation(whether -NHr* from -NH, or -CO; from -CO,H) at the interfacebetu'een surface-functionalized polyethylene and \r'ateris more difficult than that in homogeneousaqueous solution. This differenceis probably related to the low effectivedielectric constantof the interface(5=9) relativeto water (e=80). It is not known to what extentthis differencein e (and in other propertiesof the interphase,considered as a thin solventphase) is reflectedin the stabilityof the organicions relativeto their neutral forms in the interphaseand in - solution,and to whatextent in differencesin the concentrationof H' and OH in the interphaseand in solution.Self-assembled monolayers (SAMs)-especially of terminallyfunctionalized alkanethiols (HS(CH:),,X) adsorbedon gold-provide model systemswith relativelywell-ordered structures thar are useful in establishingthe fundamentalsof ionizationof protic acidsand basesat the interface betr"'eenorganic solids and water.These systems,coupled r.r'ithnew analyticalmethods such as photoacousticcalorimetry (PAC) and contact angletitration, may make it possibleto disentangle some of the complex puzzlespresented by proton-transferreactions in the environmentof the organicsolid-* ater interphase. Keyx'ords:Contact ansle titration; photoacoustic calorimetrv: poly'ethylene carboxylic acid: contact angle:polvmer surfaces; u'etting; self-assembled monolavers. -

Hydride Abstraction and Deprotonation – an Efficient Route to Low Co-Ordinate Phosphorus and Arsenic Species
Hydride Abstraction and Deprotonation – an Efficient Route to Low Co-ordinate Phosphorus and Arsenic Species Laurence J. Taylor, Michael Bühl, Piotr Wawrzyniak, Brian A. Chalmers, J. Derek Woollins, Alexandra M. Z. Slawin, Amy L. Fuller and Petr Kilian* School of Chemistry, EastCHEM, University of St Andrews, St Andrews, Fife, KY16 9ST, UK E-mail: [email protected] URL: http://chemistry.st-andrews.ac.uk/staff/pk/group/ Supporting information for this article is given via a link at the end of the document. Abstract Treatment of Acenap(PiPr2)(EH2) (Acenap = acenaphthene-5,6-diyl; 1a, E = As; 1b, E = P) with Ph3C·BF4 resulted in hydride abstraction to give [Acenap(PiPr2)(EH)][BF4] (2a, E = As; 2b, E = P). These represent the first structurally characterised phosphino/arsino-phosphonium salts with secondary arsine/phosphine groups, as well as the first example of a Lewis base stabilised primary arsenium cation. Compounds 2a and 2b were deprotonated with NaH to afford low co-ordinate species Acenap(PiPr2)(E) (3a, E = As; 3b, E = P). This provides an alternative and practical synthetic pathway to the phosphanylidene-σ4-phosphorane 3b and provides mechanistic insight into the formation of arsanylidene-σ4-phosphorane 3a, indirectly supporting the hypothesis that the previously reported dehydrogenation of 1a occurs via an ionic mechanism. Introduction 4 Alkylidene-σ -phosphoranes (R2C=PR3), more commonly known as Wittig reagents, are widely used intermediates in the formation of C=C double bonds. The arsenic and phosphorus equivalents, 4 4 arsanylidene-σ -phosphoranes (RAs=PR3) and phosphanylidene-σ -phosphoranes (RP=PR3), are rare 4 by comparison. -

Reactivity Landscape of Pyruvate Under Simulated Hydrothermal Vent
Reactivity landscape of pyruvate under simulated SEE COMMENTARY hydrothermal vent conditions Yehor Novikova and Shelley D. Copleyb,c,1 aDepartment of Chemistry and Biochemistry, bDepartment of Molecular, Cellular, and Developmental Biology, and cCooperative Institute for Research in Environmental Sciences, University of Colorado Boulder, Boulder, CO 80309 Edited by Paul G. Falkowski, Rutgers, The State University of New Jersey, New Brunswick, NJ, and approved June 14, 2013 (received for review March 14, 2013) Pyruvate is an important “hub” metabolite that is a precursor for concentrations of many components (4). Fig. 1 shows an example in amino acids, sugars, cofactors, and lipids in extant metabolic net- which the availability of catalysts for different steps in a network works. Pyruvate has been produced under simulated hydrother- results in significantly different network topologies and accumu- mal vent conditions from alkyl thiols and carbon monoxide in the lation of different products. Network topology also depends on the presence of transition metal sulfides at 250 °C [Cody GD et al. set of reagents available and the concentrations of those reagents. K (2000) Science 289(5483):1337–1340], so it is plausible that pyru- For example, the network depicted in Fig. 1 would form only and M H J M vate was formed in hydrothermal systems on the early earth. We if no were available, and would form only and if the concentration of H were very high (assuming equal rate constants report here that pyruvate reacts readily in the presence of transi- D tion metal sulfide minerals under simulated hydrothermal vent for the partitioning of between the two possible pathways). -

Introduction to Ionic Mechanisms Part I: Fundamentals of Bronsted-Lowry Acid-Base Chemistry
INTRODUCTION TO IONIC MECHANISMS PART I: FUNDAMENTALS OF BRONSTED-LOWRY ACID-BASE CHEMISTRY HYDROGEN ATOMS AND PROTONS IN ORGANIC MOLECULES - A hydrogen atom that has lost its only electron is sometimes referred to as a proton. That is because once the electron is lost, all that remains is the nucleus, which in the case of hydrogen consists of only one proton. The large majority of organic reactions, or transformations, involve breaking old bonds and forming new ones. If a covalent bond is broken heterolytically, the products are ions. In the following example, the bond between carbon and oxygen in the t-butyl alcohol molecule breaks to yield a carbocation and hydroxide ion. H3C CH3 H3C OH H3C + OH CH3 H3C A tertiary Hydroxide carbocation ion The full-headed curved arrow is being used to indicate the movement of an electron pair. In this case, the two electrons that make up the carbon-oxygen bond move towards the oxygen. The bond breaks, leaving the carbon with a positive charge, and the oxygen with a negative charge. In the absence of other factors, it is the difference in electronegativity between the two atoms that drives the direction of electron movement. When pushing arrows, remember that electrons move towards electronegative atoms, or towards areas of electron deficiency (positive, or partial positive charges). The electron pair moves towards the oxygen because it is the more electronegative of the two atoms. If we examine the outcome of heterolytic bond cleavage between oxygen and hydrogen, we see that, once again, oxygen takes the two electrons because it is the more electronegative atom. -

Organic Seminar Abstracts
L I B RA FLY OF THE U N IVERSITY OF 1LLI NOIS Q.54-1 Ii£s 1964/65 ,t.l P Return this book on or before the Latest Date stamped below. Theft, mutilation, and underlining of books are reasons for disciplinary action and may result in dismissal from the University. University of Illinois Library OCfcfcsrm L161— O-1096 Digitized by the Internet Archive in 2012 with funding from University of Illinois Urbana-Champaign http://archive.org/details/organicsemi1964651univ s ORGANIC SEMINAR ABSTRACTS 196^-65 Semester I Department of Chemistry and Chemical Engineering University of Illinois ' / SEMINAR TOPICS I Semester I96U-I965 f"/ ( Orientation in Sodium and Potassium Metalations of Aromatic Compounds Earl G. Alley 1 Structure of Cyclopropane Virgil Weiss 9 Vinylidenes and Vinylidenecarbenes Joseph C. Catlin 17 Diazene Intermediates James A. Bonham 25 Perfluoroalkyl and Polyfluoroalkyl Carbanions J. David Angerer 3^ Free Radical Additions to Allenes Raymond Feldt 43 The Structure and Biosynthesis of Quassin Richard A. Larson 52 The Decomposition of Perester Compounds Thomas Sharpe 61 The Reaction of Di-t -Butyl Peroxide with Simple Alkyl, Benzyl and Cyclic Ethers R. L. Keener 70 Rearrangements and Solvolysis in Some Allylic Systems Jack Timberlake 79 Longifolene QQ Michael A. Lintner 00 Hydrogenation ! Homogeneous Catalytic Robert Y. Ning 9° c c Total Synthesis of ( -) -Emetime R. Lambert 1* Paracyclophanes Ping C. Huang 112 Mechanism of the Thermal Rearrangement of Cyclopropane George Su 121 Some Recent Studies of the Photochemistry of Cross -Conjugated Cyclohexadienones Elizabeth McLeister 130 The Hammett Acidity Function R. P. Quirk 139 Homoaromaticity Roger A. -

APPENDIX G Acid Dissociation Constants
harxxxxx_App-G.qxd 3/8/10 1:34 PM Page AP11 APPENDIX G Acid Dissociation Constants § ϭ 0.1 M 0 ؍ (Ionic strength ( † ‡ † Name Structure* pKa Ka pKa ϫ Ϫ5 Acetic acid CH3CO2H 4.756 1.75 10 4.56 (ethanoic acid) N ϩ H3 ϫ Ϫ3 Alanine CHCH3 2.344 (CO2H) 4.53 10 2.33 ϫ Ϫ10 9.868 (NH3) 1.36 10 9.71 CO2H ϩ Ϫ5 Aminobenzene NH3 4.601 2.51 ϫ 10 4.64 (aniline) ϪO SNϩ Ϫ4 4-Aminobenzenesulfonic acid 3 H3 3.232 5.86 ϫ 10 3.01 (sulfanilic acid) ϩ NH3 ϫ Ϫ3 2-Aminobenzoic acid 2.08 (CO2H) 8.3 10 2.01 ϫ Ϫ5 (anthranilic acid) 4.96 (NH3) 1.10 10 4.78 CO2H ϩ 2-Aminoethanethiol HSCH2CH2NH3 —— 8.21 (SH) (2-mercaptoethylamine) —— 10.73 (NH3) ϩ ϫ Ϫ10 2-Aminoethanol HOCH2CH2NH3 9.498 3.18 10 9.52 (ethanolamine) O H ϫ Ϫ5 4.70 (NH3) (20°) 2.0 10 4.74 2-Aminophenol Ϫ 9.97 (OH) (20°) 1.05 ϫ 10 10 9.87 ϩ NH3 ϩ ϫ Ϫ10 Ammonia NH4 9.245 5.69 10 9.26 N ϩ H3 N ϩ H2 ϫ Ϫ2 1.823 (CO2H) 1.50 10 2.03 CHCH CH CH NHC ϫ Ϫ9 Arginine 2 2 2 8.991 (NH3) 1.02 10 9.00 NH —— (NH2) —— (12.1) CO2H 2 O Ϫ 2.24 5.8 ϫ 10 3 2.15 Ϫ Arsenic acid HO As OH 6.96 1.10 ϫ 10 7 6.65 Ϫ (hydrogen arsenate) (11.50) 3.2 ϫ 10 12 (11.18) OH ϫ Ϫ10 Arsenious acid As(OH)3 9.29 5.1 10 9.14 (hydrogen arsenite) N ϩ O H3 Asparagine CHCH2CNH2 —— —— 2.16 (CO2H) —— —— 8.73 (NH3) CO2H *Each acid is written in its protonated form. -

Infrared Spectroscopy of Protonated Acetic Acid
Probing Elusive Cations: Infrared Spectroscopy of Protonated Acetic Acid Julia A. Davies,a) Nicholas A. Besley,b) Shengfu Yanga) and Andrew M. Ellisa),* a) Department of Chemistry, University of Leicester, University Road, Leicester, LE1 7RH, UK b) School of Chemistry, University of Nottingham, University Park, Nottingham, NG7 2RD, UK *Corresponding author: Email: [email protected] Manuscript submitted to The Journal of Physical Chemistry Letters 1 Abstract Protonated carboxylic acids, (RCOOH)H+, are the initial intermediates in acid-catalyzed (Fischer) esterification reactions. However the identity of the isomeric form is under debate. Surprisingly, no optical spectra have been reported for any isomer of the protonated carboxylic acid monomer, despite it being a fundamental organic cation. Here, we address these issues by using a new approach to prepare cold He-tagged cations of protonated acetic acid (AA), which entails electron ionization of helium nanodroplets containing metastable dimers of AA. The protonated species is subsequently probed using infrared photodissociation spectroscopy and, following a comparison with calculations, we identify the two isomers whose roles are debated in Fischer esterification. These are the carbonyl-protonated E,Z isomer and the metastable hydroxyl-protonated isomer. Our technique provides a novel approach that can be applied to other elusive ionic species. TOC Graphic 2 The mechanism of the acid-catalyzed (Fischer) esterification of carboxylic acids was first explored in detail in the 1930s.1,2 This early mechanistic work suggested that initial protonation occurs at the hydroxyl oxygen atom. In the case of an acetic acid monomer (AA), this leads to formation of the structure labelled as prot-OH in Figure 1.