Intact Carbonic Acid Is a Viable Protonating Agent for Biological Bases
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Reactivity Landscape of Pyruvate Under Simulated Hydrothermal Vent
Reactivity landscape of pyruvate under simulated SEE COMMENTARY hydrothermal vent conditions Yehor Novikova and Shelley D. Copleyb,c,1 aDepartment of Chemistry and Biochemistry, bDepartment of Molecular, Cellular, and Developmental Biology, and cCooperative Institute for Research in Environmental Sciences, University of Colorado Boulder, Boulder, CO 80309 Edited by Paul G. Falkowski, Rutgers, The State University of New Jersey, New Brunswick, NJ, and approved June 14, 2013 (received for review March 14, 2013) Pyruvate is an important “hub” metabolite that is a precursor for concentrations of many components (4). Fig. 1 shows an example in amino acids, sugars, cofactors, and lipids in extant metabolic net- which the availability of catalysts for different steps in a network works. Pyruvate has been produced under simulated hydrother- results in significantly different network topologies and accumu- mal vent conditions from alkyl thiols and carbon monoxide in the lation of different products. Network topology also depends on the presence of transition metal sulfides at 250 °C [Cody GD et al. set of reagents available and the concentrations of those reagents. K (2000) Science 289(5483):1337–1340], so it is plausible that pyru- For example, the network depicted in Fig. 1 would form only and M H J M vate was formed in hydrothermal systems on the early earth. We if no were available, and would form only and if the concentration of H were very high (assuming equal rate constants report here that pyruvate reacts readily in the presence of transi- D tion metal sulfide minerals under simulated hydrothermal vent for the partitioning of between the two possible pathways). -

Introduction to Ionic Mechanisms Part I: Fundamentals of Bronsted-Lowry Acid-Base Chemistry
INTRODUCTION TO IONIC MECHANISMS PART I: FUNDAMENTALS OF BRONSTED-LOWRY ACID-BASE CHEMISTRY HYDROGEN ATOMS AND PROTONS IN ORGANIC MOLECULES - A hydrogen atom that has lost its only electron is sometimes referred to as a proton. That is because once the electron is lost, all that remains is the nucleus, which in the case of hydrogen consists of only one proton. The large majority of organic reactions, or transformations, involve breaking old bonds and forming new ones. If a covalent bond is broken heterolytically, the products are ions. In the following example, the bond between carbon and oxygen in the t-butyl alcohol molecule breaks to yield a carbocation and hydroxide ion. H3C CH3 H3C OH H3C + OH CH3 H3C A tertiary Hydroxide carbocation ion The full-headed curved arrow is being used to indicate the movement of an electron pair. In this case, the two electrons that make up the carbon-oxygen bond move towards the oxygen. The bond breaks, leaving the carbon with a positive charge, and the oxygen with a negative charge. In the absence of other factors, it is the difference in electronegativity between the two atoms that drives the direction of electron movement. When pushing arrows, remember that electrons move towards electronegative atoms, or towards areas of electron deficiency (positive, or partial positive charges). The electron pair moves towards the oxygen because it is the more electronegative of the two atoms. If we examine the outcome of heterolytic bond cleavage between oxygen and hydrogen, we see that, once again, oxygen takes the two electrons because it is the more electronegative atom. -

Infrared Spectroscopy of Protonated Acetic Acid
Probing Elusive Cations: Infrared Spectroscopy of Protonated Acetic Acid Julia A. Davies,a) Nicholas A. Besley,b) Shengfu Yanga) and Andrew M. Ellisa),* a) Department of Chemistry, University of Leicester, University Road, Leicester, LE1 7RH, UK b) School of Chemistry, University of Nottingham, University Park, Nottingham, NG7 2RD, UK *Corresponding author: Email: [email protected] Manuscript submitted to The Journal of Physical Chemistry Letters 1 Abstract Protonated carboxylic acids, (RCOOH)H+, are the initial intermediates in acid-catalyzed (Fischer) esterification reactions. However the identity of the isomeric form is under debate. Surprisingly, no optical spectra have been reported for any isomer of the protonated carboxylic acid monomer, despite it being a fundamental organic cation. Here, we address these issues by using a new approach to prepare cold He-tagged cations of protonated acetic acid (AA), which entails electron ionization of helium nanodroplets containing metastable dimers of AA. The protonated species is subsequently probed using infrared photodissociation spectroscopy and, following a comparison with calculations, we identify the two isomers whose roles are debated in Fischer esterification. These are the carbonyl-protonated E,Z isomer and the metastable hydroxyl-protonated isomer. Our technique provides a novel approach that can be applied to other elusive ionic species. TOC Graphic 2 The mechanism of the acid-catalyzed (Fischer) esterification of carboxylic acids was first explored in detail in the 1930s.1,2 This early mechanistic work suggested that initial protonation occurs at the hydroxyl oxygen atom. In the case of an acetic acid monomer (AA), this leads to formation of the structure labelled as prot-OH in Figure 1. -

Protonation Patterns in Reduced and Oxidized Forms of Electron Transfer Proteins
Protonation patterns in reduced and oxidized forms of electron transfer proteins Dissertation For the award of the degree “doctor rerum naturalium” Division of Mathematics and Natural Sciences of the Georg-August-Universit¨at G¨ottingen submitted by Plamen Dobrev from Harmanli G¨ottingen 2012 Prof. Dr. Helmut Grubm¨uller (Reviewer) Department of Theoretical and Computational Biophysics, Max Planck In- stitute for Biophysical Chemistry G¨ottingen Prof. Dr. Marcus M¨uller (Reviewer) Institute for Theoretical Physics, Georg August University G¨ottingen Prof. Dr. Claudia Steinem Institute for Organic and Biomolecular Chemistry, Georg August University G¨ottingen Date of the oral examination: 08.05.2012 1 It is declared that the presented thesis has been written independently and with no other sources and aids than quoted. G¨ottingen, 10.04.2012 Plamen Dobrev 2 Contents 1 Introduction 6 2 Theory and Methods 13 2.1 Moleculardynamics........................ 13 2.2 pKa calculations of an acid in solution . 15 2.3 Free Energy calculation of deprotonation of an amino acid in MDsimulation .......................... 17 2.4 pKa calculation of ionizable groups in proteins using constant pHMD .............................. 19 2.5 Construction of the model compounds and the titratable amino acidsintheprotein ........................ 23 2.5.1 Residues with Carboxyl titratable group . 24 2.5.2 Histidine . 26 2.5.3 Tyrosine.......................... 27 2.5.4 Modelcompounds.. .. 27 2.6 Keeping the simulation box neutral upon protonation or de- protonation ............................ 29 2.6.1 Thechangeoftheionicstrength. 29 2.6.2 Restraing the coupled water molecules and the entropy change due tothe restraining potential . 30 3 2.6.3 Distance between the titrating site and the coupled watermolecule ..................... -

Methylation Deficiency Disrupts Biological Rhythms from Bacteria To
ARTICLE https://doi.org/10.1038/s42003-020-0942-0 OPEN Methylation deficiency disrupts biological rhythms from bacteria to humans ✉ Jean-Michel Fustin 1,16,18 , Shiqi Ye1,18, Christin Rakers 2, Kensuke Kaneko3, Kazuki Fukumoto1, Mayu Yamano1, Marijke Versteven4, Ellen Grünewald5, Samantha J. Cargill5, T. Katherine Tamai 6, Yao Xu7, Maria Luísa Jabbur7, Rika Kojima8, Melisa L. Lamberti9, Kumiko Yoshioka-Kobayashi10, David Whitmore 11, 1234567890():,; Stephanie Tammam12, P. Lynne Howell 12,13, Ryoichiro Kageyama10, Takuya Matsuo14, Ralf Stanewsky 4, Diego A. Golombek9, Carl Hirschie Johnson7, Hideaki Kakeya 3, Gerben van Ooijen 5 & ✉ Hitoshi Okamura15,17 The methyl cycle is a universal metabolic pathway providing methyl groups for the methy- lation of nuclei acids and proteins, regulating all aspects of cellular physiology. We have previously shown that methyl cycle inhibition in mammals strongly affects circadian rhythms. Since the methyl cycle and circadian clocks have evolved early during evolution and operate in organisms across the tree of life, we sought to determine whether the link between the two is also conserved. Here, we show that methyl cycle inhibition affects biological rhythms in species ranging from unicellular algae to humans, separated by more than 1 billion years of evolution. In contrast, the cyanobacterial clock is resistant to methyl cycle inhibition, although we demonstrate that methylations themselves regulate circadian rhythms in this organism. Mammalian cells with a rewired bacteria-like methyl cycle are protected, like cyanobacteria, from methyl cycle inhibition, providing interesting new possibilities for the treatment of methylation deficiencies. 1 Graduate School of Pharmaceutical Sciences, Laboratory of Molecular Metabology, Kyoto University, Kyoto, Japan. -

The Role of Protonation States in Ligand-Receptor Recognition and Binding Marharyta Petukh Clemson University
Clemson University TigerPrints Publications Physics and Astronomy 5-2013 The Role of Protonation States in Ligand-Receptor Recognition and Binding Marharyta Petukh Clemson University Shannon Stefl Clemson University Emil Alexov Clemson University, [email protected] Follow this and additional works at: https://tigerprints.clemson.edu/physastro_pubs Part of the Biological and Chemical Physics Commons Recommended Citation Please use publisher's recommended citation. This Article is brought to you for free and open access by the Physics and Astronomy at TigerPrints. It has been accepted for inclusion in Publications by an authorized administrator of TigerPrints. For more information, please contact [email protected]. NIH Public Access Author Manuscript Curr Pharm Des. Author manuscript; available in PMC 2013 May 30. NIH-PA Author ManuscriptPublished NIH-PA Author Manuscript in final edited NIH-PA Author Manuscript form as: Curr Pharm Des. 2013 ; 19(23): 4182–4190. The Role of Protonation States in Ligand-Receptor Recognition and Binding Marharyta Petukh1, Shannon Stefl1, and Emil Alexov1 1Computational Biophysics and Bioinformatics, Department of Physics and Astronomy, Clemson University, Clemson, SC 29634, USA Abstract In this review we discuss the role of protonation states in receptor-ligand interactions, providing experimental evidences and computational predictions that complex formation may involve titratable groups with unusual pKa’s and that protonation states frequently change from unbound to bound states. These protonation changes result in proton uptake/release, which in turn causes the pH-dependence of the binding. Indeed, experimental data strongly suggests that almost any binding is pH-dependent and to be correctly modeled, the protonation states must be properly assigned prior to and after the binding. -

Chemical Science
Chemical Science View Article Online EDGE ARTICLE View Journal | View Issue A mechanistic study of proton reduction catalyzed by a pentapyridine cobalt complex: evidence for Chem. Sci. Cite this: , 2013, 4, 1578 involvement of an anation-based pathway† Amanda E. King,a Yogesh Surendranath,a Nicholas A. Piro,ae Julian P. Bigi,ae Jeffrey R. Long*ad and Christopher J. Chang*abce 2+ The pentapyridine cobalt complex [Co(PY5Me2)] and its congeners have been shown to catalyze proton reduction to hydrogen in aqueous solution over a wide pH range using electrical or solar energy input. Here, we employ electrochemical and spectroscopic studies to examine the mechanisms of proton reduction by this parent complex under soluble, diffusion-limited conditions in acetonitrile with acetic acid as the proton donor. Two pathways for proton reduction are identified via cyclic voltammetry: one pathway occurring from an acetonitrile-bound CoII/I couple and the other pathway operating from an acetate-bound CoII/I couple. Kinetics studies support protonation of a CoI species as the rate- determining step for both processes, and additional electrochemical measurements further suggest that the onset of catalysis from the acetonitrile-bound CoII/I couple is highly affected by catalyst electronics. Received 14th December 2012 Taken together, this work not only establishes the CoPY5Me unit as a unique molecular platform that Accepted 12th February 2013 2 catalyzes the reduction of protons under soluble, diffusion-limited conditions in both aqueous and DOI: 10.1039/c3sc22239j organic media, but also highlights the participation of anation processes that are likely relevant for a www.rsc.org/chemicalscience wide range of hydrogen-producing and related catalytic systems. -

Ion Stability of Nucleic Acids in Infrared Matrix-Assisted Laser Desorption/Ionization Mass Spectrometry
Nucleic Acids Research, 1993, Vol. 21, No. 15 3347-3357 Ion stability of nucleic acids in infrared matrix-assisted laser desorption/ionization mass spectrometry E.Nordhoff, R.Cramer, M.Karas, F.Hillenkamp, F.Kirpekar1, K.Kristiansen1 and P.Roepstorffl Institute for Medical Physics and Biophysics, University of Munster, Robert-Koch-Strasse 31, 4400 Munster, Germany and 1Department of Molecular Biology, University of Odense, Campusvej 55, 5230 Odense M., Denmark Received May 28, 1993; Accepted June 21, 1993 ABSTRACT Matrix-assisted laser desorption/ionization mass suitable matrix of molecules having a strong absorption at the spectrometry (MALDI-MS) with infrared laser light of a laser wavelength and present in high molar excess over the wavelength of 2.94 zm has been used for the analysis analyte. This induces an efficient transfer ofthe laser-pulse energy of nucleic acids. Spectra of oligodeoxynucleotides up to the analyte and results in a soft desorption process. It is to 26 nucleotides, oligothymidylic acids up to 100 suggested that the matrix molecules also play a role in the nucleotides as well as different synthetic RNA ionization of the analyte molecules.9 Different lasers and oligomers and RNA transcripts up to 104 nucleotides wavelengths in the UV (mainly 266 nm, 337 nm and 355 nm) are presented. A main problem in the analysis of and IR (mainly 2.94 ,um, 2.79 ym and 10.6Am) have been used oligodeoxynucleotides was found to be related to the for this technique. Complex mixtures of proteins and peptides loss of bases. The stability of oligothymidylic acids as are routinely mass analysed. -

UV Matrix-Assisted Laser Desorption Ionization: Principles, Instrumentation, and Applications Often Used to Promote the Drying Process
Desorption by Photons UV Matrix-Assisted Laser Desorption often discussed separately (see Sections 4 and 5), one needs to be reminded that many aspects of both Ionization: Principles, Instrumentation, and processes are coupled and occur at the same time. Applications Moreover, the exact desorption/ionization pathways critically depend on the material properties of the compounds and on the laser irradiation parameters. 1. Introduction The lasers most often used for MALDI-MS are pulsed ultraviolet (UV) lasers with wavelengths close Matrix-assisted laser desorption ionization (MALDI) to the maximum UV absorption of the matrix (e.g., is, together with electrospray ionization (ESI) (see 337 nm, nitrogen gas laser; and 355 nm, frequency- Chapter 7 (this volume): Electrospray Ionization: tripled Nd:YAG solid state laser) and pulse durations Principles and Instrumentation ), one of the most of 1–10 ns. Other lasers and laser wavelengths can be widely employed ionization techniques in biological used for MALDI-MS but less frequently owing to mass spectrometry (MS). MALDI was developed in generally inferior MALDI and/or laser performance the second half of the 1980s ( 1,2 ), and its first pub- at these wavelengths. Hence, UV-MALDI at the lished applications coincided with the first reports of above wavelengths is the predominant MALDI ESI analyses of proteins. Owing to a low degree of method in commercially available MALDI-MS in- analyte fragmentation, MALDI and ESI are often strumentation and in the main application areas of referred to as ‘‘soft’’ ionization techniques, and as biological mass spectrometry (e.g., in proteomics: see such both paved the way for one half of the 2002 Chapter 1 (Volume 2): Proteomies: An Overview ). -

Green Rust: the Simple Organizing ‘Seed’ of All Life?
life Review Green Rust: The Simple Organizing ‘Seed’ of All Life? Michael J. Russell Planetary Chemistry and Astrobiology, Jet Propulsion Laboratory, California Institute of Technology, Pasadena, CA 91109-8099, USA; [email protected] Received: 6 June 2018; Accepted: 14 August 2018; Published: 27 August 2018 Abstract: Korenaga and coworkers presented evidence to suggest that the Earth’s mantle was dry and water filled the ocean to twice its present volume 4.3 billion years ago. Carbon dioxide was constantly exhaled during the mafic to ultramafic volcanic activity associated with magmatic plumes that produced the thick, dense, and relatively stable oceanic crust. In that setting, two distinct and major types of sub-marine hydrothermal vents were active: ~400 ◦C acidic springs, whose effluents bore vast quantities of iron into the ocean, and ~120 ◦C, highly alkaline, and reduced vents exhaling from the cooler, serpentinizing crust some distance from the heads of the plumes. When encountering the alkaline effluents, the iron from the plume head vents precipitated out, forming mounds likely surrounded by voluminous exhalative deposits similar to the banded iron formations known from the Archean. These mounds and the surrounding sediments, comprised micro or nano-crysts of the variable valence FeII/FeIII oxyhydroxide known as green rust. The precipitation of green rust, along with subsidiary iron sulfides and minor concentrations of nickel, cobalt, and molybdenum in the environment at the alkaline springs, may have established both the key bio-syntonic disequilibria and the means to properly make use of them—the elements needed to effect the essential inanimate-to-animate transitions that launched life. -

Direct Determination of Protonation States and Visualization of Hydrogen Bonding in a Glycoside Hydrolase with Neutron Crystallography
Direct determination of protonation states and visualization of hydrogen bonding in a glycoside hydrolase with neutron crystallography Qun Wana, Jerry M. Parksb, B. Leif Hansonc, Suzanne Zoe Fisherd, Andreas Ostermanne, Tobias E. Schraderf, David E. Grahamg, Leighton Coatesh, Paul Langanh, and Andrey Kovalevskyh,1 aDepartment of Physics, College of Science, Nanjing Agricultural University, Nanjing 210095, People’s Republic of China; bUniversity of Tennessee/Oak Ridge National Laboratory Center for Molecular Biophysics, Biosciences Division, Oak Ridge National Laboratory, Oak Ridge, TN 37831; cChemistry Department, University of Toledo, Toledo, OH 43606; dScientific Activities Division, European Spallation Source, Lund 22100, Sweden; eHeinz Maier-Leibnitz Zentrum, Technische Universität München, 85748 Garching, Germany; fJülich Centre for Neutron Science at Heinz Maier-Leibnitz Zentrum, Forschungszentrum Jülich GmbH, 85747 Garching, Germany; gBiosciences Division, Oak Ridge National Laboratory, Oak Ridge, TN 37831; and hBiology and Soft Matter Division, Oak Ridge National Laboratory, Oak Ridge, TN 37831 Edited by Dagmar Ringe, Brandeis University, Waltham, MA, and accepted by the Editorial Board August 18, 2015 (received for review March 12, 2015) Glycoside hydrolase (GH) enzymes apply acid/base chemistry to oxygen in the rate-limiting step (8–10). Two models for the cata- catalyze the decomposition of complex carbohydrates. These lytic mechanism of β-glycoside hydrolases have been proposed: ubiquitous enzymes accept protons from solvent and donate them (i) a double displacement pathway first proposed by Koshland, to substrates at close to neutral pH by modulating the pKa values essentially proceeding as an SN2 reaction (11, 12) and (ii)an of key side chains during catalysis. However, it is not known how oxocarbenium ion intermediate pathway originally proposed by the catalytic acid residue acquires a proton and transfers it effi- Phillips (13), occurring as an SN1 reaction (Fig. -
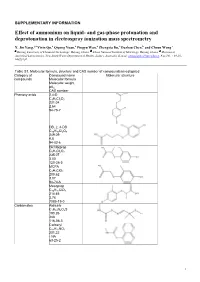
Effect of Ammonium on Liquid- and Gas-Phase Protonation and Deprotonation in Electrospray Ionization Mass Spectrometry
SUPPLEMENTARY INFORMATION Effect of ammonium on liquid- and gas-phase protonation and deprotonation in electrospray ionization mass spectrometry X. Jin Yang,*a Yixin Qu,a Qipeng Yuan,a Pingyu Wan,a Zhengxia Du,a Dazhou Chen,b and Choon Wong c a b c Beijing University of Chemical Technology, Beijing, China; China National Institute of Metrology, Beijing, China; Division of Analytical Laboratories, New South Wales Department of Health, Sydney, Australia. E-mail: [email protected]. Fax/Tel: +86-10- 64421030. Table S1. Molecular formula, structure and CAS number of compounds investigated Category of Compound name Molecular structure compounds Molecular formula Molecular weight pKa CAS number Phenoxy acids 2,4-D C8H6Cl2O3 221.04 2.64 94-75-7 DB, 2, 4-DB C10H10Cl2O3 249.09 4.8 94-82-6 Dichlorprop C9H8Cl2O3 235.07 3.00 120-36-5 MCPA C9H9ClO3 200.62 3.07 94-74-6 Mecoprop C10H11ClO3 214.65 3.78 7085-19-0 Carbamates Aldicarb C7H14N2O2S 190.26 -NA 116-06-3 Carbaryl C12H11NO2 201.22 - NA 63-25-2 1 Carbendazim C9H9N3O2 191.19 4.2 10605-21-7 Carbofuran C12H15NO3 221.26 - NA 1563-66-2 Imidazole Prochloraz C15H16Cl3N3O2 376.67 3.8 67747-09-5 Organophosphate Dimethoate C5H12NO3PS2 229.25 2.0 (20oC) 60-51-5 Pirimiphos-ethyl C13H24N3O3PS 333.38 4.30 23505-41-1 Thicarbamate Molinate (Liquid) C9H17NOS 187.30 -NA 2212-67-1 Sulfonylurea Bensulfuron methyl C16H18N4O7S 410.40 5.2 83055-99-6 Chlorsulfuron C12H12ClN5O4S 357.77 3.60 64902-72-3 Metsulfuron methyl C14H15N5O6S 381.36 3.33 74223-64-6 2 Rimsulfuron C14H17N5O7S2 431.44 4 122931-48-0 Sulfometuron