In Utero Exposure to Chlordecone Affects Histone Modifications And
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Iron Depletion Reduces Abce1 Transcripts While Inducing The
Preprints (www.preprints.org) | NOT PEER-REVIEWED | Posted: 22 October 2019 doi:10.20944/preprints201910.0252.v1 1 Research Article 2 Iron depletion Reduces Abce1 Transcripts While 3 Inducing the Mitophagy Factors Pink1 and Parkin 4 Jana Key 1,2, Nesli Ece Sen 1, Aleksandar Arsovic 1, Stella Krämer 1, Robert Hülse 1, Suzana 5 Gispert-Sanchez 1 and Georg Auburger 1,* 6 1 Experimental Neurology, Goethe University Medical School, 60590 Frankfurt am Main; 7 2 Faculty of Biosciences, Goethe-University Frankfurt am Main, Germany 8 * Correspondence: [email protected] 9 10 Abstract: Lifespan extension was recently achieved in Caenorhabditis elegans nematodes by 11 mitochondrial stress and mitophagy, triggered via iron depletion. Conversely in man, deficient 12 mitophagy due to Pink1/Parkin mutations triggers iron accumulation in patient brain and limits 13 survival. We now aimed to identify murine fibroblast factors, which adapt their mRNA expression 14 to acute iron manipulation, relate to mitochondrial dysfunction and may influence survival. After 15 iron depletion, expression of the plasma membrane receptor Tfrc with its activator Ireb2, the 16 mitochondrial membrane transporter Abcb10, the heme-release factor Pgrmc1, the heme- 17 degradation enzyme Hmox1, the heme-binding cholesterol metabolizer Cyp46a1, as well as the 18 mitophagy regulators Pink1 and Parkin showed a negative correlation to iron levels. After iron 19 overload, these factors did not change expression. Conversely, a positive correlation of mRNA levels 20 with both conditions of iron availability was observed for the endosomal factors Slc11a2 and Steap2, 21 as well as for the iron-sulfur-cluster (ISC)-containing factors Ppat, Bdh2 and Nthl1. -
Supplementary Text and Figures.Pdf
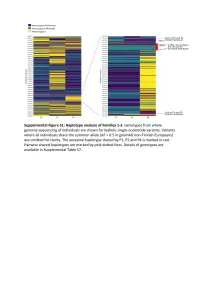
Supplemental Figure S2. PRIM1 immunoblots (20-75 kDa). (A) Corresponds to Figure 2D, with additional bands seen in P2 at 75 kDa and 25 kDa. (B) Independent experiment with cell lysates from C and P2 in which additional bands at 25 kDa not evident. (C) Corresponds to Figure 2D. No additional band visible at 25 kDa. (D) Validation of monoclonal antibody (8G10) with siRNA to PRIM1 in HeLa cells. siLUC, luciferase negative control. siPRIM1, siRNA targeting PRIM1 transcript. Vinculin used as a loading control. Supplemental Figure S3: Predicted destabilisation of PRIM1 by the C301R substitution. (A) Cysteine 301 lies in a buried hydrophobic region in PRIM1. DNA primase dimer crystal structure (PDB: 4BPU). PRIM1 residues shaded according to solvent accessibility. Cysteine 301, substituted to arginine in P5, lies in a buried hydrophobic region. (B) Cys301 is evolutionarily conserved in vertebrates. Neither arginine or other large or charged amino acids are observed at this position in other species. (C) The C301R substitution observed in P5 is predicted to lead to destabilisation of all available PRIM1 crystal structures. In contrast, substitution with leucine or threonine, as observed in some orthologous proteins, is not predicted to lead to destabilisation. Data points, predicted changes in free-energy (ΔΔG) from FoldX plotted for each of 14 available crystal structures. (D) Immunoblotting of RPE1 cells transfected with dual reporter constructs (as described in Figure 3B and Materials and Methods) confirms production of PRIM1-GFP and FLAG-SR at expected molecular weights and shows reduced protein levels for the C301R variant. n=2 experiments shown. C301R protein levels relative to WT after normalization to actin loading control indicated underneath the blot. -

Bioinformatics-Based Screening of Key Genes for Transformation of Liver
Jiang et al. J Transl Med (2020) 18:40 https://doi.org/10.1186/s12967-020-02229-8 Journal of Translational Medicine RESEARCH Open Access Bioinformatics-based screening of key genes for transformation of liver cirrhosis to hepatocellular carcinoma Chen Hao Jiang1,2, Xin Yuan1,2, Jiang Fen Li1,2, Yu Fang Xie1,2, An Zhi Zhang1,2, Xue Li Wang1,2, Lan Yang1,2, Chun Xia Liu1,2, Wei Hua Liang1,2, Li Juan Pang1,2, Hong Zou1,2, Xiao Bin Cui1,2, Xi Hua Shen1,2, Yan Qi1,2, Jin Fang Jiang1,2, Wen Yi Gu4, Feng Li1,2,3 and Jian Ming Hu1,2* Abstract Background: Hepatocellular carcinoma (HCC) is the most common type of liver tumour, and is closely related to liver cirrhosis. Previous studies have focussed on the pathogenesis of liver cirrhosis developing into HCC, but the molecular mechanism remains unclear. The aims of the present study were to identify key genes related to the transformation of cirrhosis into HCC, and explore the associated molecular mechanisms. Methods: GSE89377, GSE17548, GSE63898 and GSE54236 mRNA microarray datasets from Gene Expression Omni- bus (GEO) were analysed to obtain diferentially expressed genes (DEGs) between HCC and liver cirrhosis tissues, and network analysis of protein–protein interactions (PPIs) was carried out. String and Cytoscape were used to analyse modules and identify hub genes, Kaplan–Meier Plotter and Oncomine databases were used to explore relationships between hub genes and disease occurrence, development and prognosis of HCC, and the molecular mechanism of the main hub gene was probed using Kyoto Encyclopedia of Genes and Genomes(KEGG) pathway analysis. -

Gene Expression Profiling Analysis Contributes to Understanding the Association Between Non-Syndromic Cleft Lip and Palate, and Cancer
2110 MOLECULAR MEDICINE REPORTS 13: 2110-2116, 2016 Gene expression profiling analysis contributes to understanding the association between non-syndromic cleft lip and palate, and cancer HONGYI WANG, TAO QIU, JIE SHI, JIULONG LIANG, YANG WANG, LIANGLIANG QUAN, YU ZHANG, QIAN ZHANG and KAI TAO Department of Plastic Surgery, General Hospital of Shenyang Military Area Command, PLA, Shenyang, Liaoning 110016, P.R. China Received March 10, 2015; Accepted December 18, 2015 DOI: 10.3892/mmr.2016.4802 Abstract. The present study aimed to investigate the for NSCL/P were implicated predominantly in the TGF-β molecular mechanisms underlying non-syndromic cleft lip, signaling pathway, the cell cycle and in viral carcinogenesis. with or without cleft palate (NSCL/P), and the association The TP53, CDK1, SMAD3, PIK3R1 and CASP3 genes were between this disease and cancer. The GSE42589 data set found to be associated, not only with NSCL/P, but also with was downloaded from the Gene Expression Omnibus data- cancer. These results may contribute to a better understanding base, and contained seven dental pulp stem cell samples of the molecular mechanisms of NSCL/P. from children with NSCL/P in the exfoliation period, and six controls. Differentially expressed genes (DEGs) were Introduction screened using the RankProd method, and their potential functions were revealed by pathway enrichment analysis and Non-syndromic cleft lip, with or without cleft palate (NSCL/P) construction of a pathway interaction network. Subsequently, is one of the most common types of congenital defect and cancer genes were obtained from six cancer databases, and affects 3.4-22.9/10,000 individuals worldwide (1). -

PRIM1 Polyclonal ANTIBODY Catalog Number:10773-1-AP 2 Publications
For Research Use Only PRIM1 Polyclonal ANTIBODY www.ptglab.com Catalog Number:10773-1-AP 2 Publications Catalog Number: GenBank Accession Number: Purification Method: Basic Information 10773-1-AP BC005266 Antigen affinity purification Size: GeneID (NCBI): Recommended Dilutions: 150UL , Concentration: 133 μg/ml by 5557 WB 1:500-1:1000 Bradford method using BSA as the Full Name: IP 0.5-4.0 ug for IP and 1:200-1:1000 standard; primase, DNA, polypeptide 1 (49kDa) for WB Source: IHC 1:20-1:200 Calculated MW: IF 1:50-1:500 Rabbit 50 kDa Isotype: Observed MW: IgG 50 kDa Immunogen Catalog Number: AG1124 Applications Tested Applications: Positive Controls: IF, IHC, IP, WB, ELISA WB : K-562 cells, HeLa cells, Sp2/0 cells Cited Applications: IP : HeLa cells, IHC IHC : human lymphoma tissue, Species Specificity: human, mouse, rat IF : HeLa cells, Cited Species: mouse Note-IHC: suggested antigen retrieval with TE buffer pH 9.0; (*) Alternatively, antigen retrieval may be performed with citrate buffer pH 6.0 DNA replication in human cells is initiated by a complex apparatus containing a DNA polymerase-alpha/primase Background Information complex. Primase synthesizes oligoribonucleotides that serve as primers for the initiation of DNA synthesis. It plays a role in both the initiation of DNA replication and the synthesis of Okazaki fragments for lagging strand synthesis. PRIM1 is a subnuit of this complex. Notable Publications Author Pubmed ID Journal Application Pierre Kunz 30986223 PLoS One IHC Liam C Hunt 31365869 Cell Rep Storage: Storage Store at -20°C. Stable for one year after shipment. -

Consequences and Resolution of Transcription–Replication Conflicts
life Review Consequences and Resolution of Transcription–Replication Conflicts Maxime Lalonde †, Manuel Trauner †, Marcel Werner † and Stephan Hamperl * Institute of Epigenetics and Stem Cells (IES), Helmholtz Zentrum München, 81377 Munich, Germany; [email protected] (M.L.); [email protected] (M.T.); [email protected] (M.W.) * Correspondence: [email protected] † These authors contributed equally. Abstract: Transcription–replication conflicts occur when the two critical cellular machineries respon- sible for gene expression and genome duplication collide with each other on the same genomic location. Although both prokaryotic and eukaryotic cells have evolved multiple mechanisms to coordinate these processes on individual chromosomes, it is now clear that conflicts can arise due to aberrant transcription regulation and premature proliferation, leading to DNA replication stress and genomic instability. As both are considered hallmarks of aging and human diseases such as cancer, understanding the cellular consequences of conflicts is of paramount importance. In this article, we summarize our current knowledge on where and when collisions occur and how these en- counters affect the genome and chromatin landscape of cells. Finally, we conclude with the different cellular pathways and multiple mechanisms that cells have put in place at conflict sites to ensure the resolution of conflicts and accurate genome duplication. Citation: Lalonde, M.; Trauner, M.; Keywords: transcription–replication conflicts; genomic instability; R-loops; torsional stress; common Werner, M.; Hamperl, S. fragile sites; early replicating fragile sites; replication stress; chromatin; fork reversal; MIDAS; G-MiDS Consequences and Resolution of Transcription–Replication Conflicts. Life 2021, 11, 637. https://doi.org/ 10.3390/life11070637 1. -

R‐Loop Formation During S Phase Is Restricted by Primpol‐Mediated
Published online: November 26, 2018 Article R-loop formation during S phase is restricted by PrimPol-mediated repriming Sasa Svikovic1, Alastair Crisp1, Sue Mei Tan-Wong2, Thomas A Guilliam3, Aidan J Doherty3, Nicholas J Proudfoot2, Guillaume Guilbaud1 & Julian E Sale1,* Abstract sequences pose a barrier to DNA synthesis in vivo and the conse- quences of their doing so are not well understood. During DNA replication, conflicts with ongoing transcription are It is well established that long repetitive tracts lead to problems frequent and require careful management to avoid genetic insta- with both replication and transcription. For example, a long tract of bility. R-loops, three-stranded nucleic acid structures comprising a polypurine–polypyrimidine (GAA)n repeats (in which n can exceed DNA:RNA hybrid and displaced single-stranded DNA, are important 1,500) is linked to the inherited neurodegenerative disorder Friedre- drivers of damage arising from such conflicts. How R-loops stall ich’s ataxia (Campuzano et al, 1996). These repeats can form H- replication and the mechanisms that restrain their formation DNA (Frank-Kamenetskii & Mirkin, 1995), a triplex DNA structure during S phase are incompletely understood. Here, we show in vivo able to block replication both in bacterial, yeast and human cells how R-loop formation drives a short purine-rich repeat, (GAA)10,to (Ohshima et al, 1998; Krasilnikova & Mirkin, 2004; Chandok et al, become a replication impediment that engages the repriming 2012), which can promote genetic instability of the repeat (Gerhardt activity of the primase-polymerase PrimPol. Further, the absence et al, 2016). Furthermore, these repetitive tracts are prone to form of PrimPol leads to significantly increased R-loop formation around R-loops (Groh et al, 2014), three-stranded nucleic acid structures in this repeat during S phase. -

DNA Polymerases at the Eukaryotic Replication Fork Thirty Years After: Connection to Cancer
cancers Review DNA Polymerases at the Eukaryotic Replication Fork Thirty Years after: Connection to Cancer Youri I. Pavlov 1,2,* , Anna S. Zhuk 3 and Elena I. Stepchenkova 2,4 1 Eppley Institute for Research in Cancer and Allied Diseases and Buffett Cancer Center, University of Nebraska Medical Center, Omaha, NE 68198, USA 2 Department of Genetics and Biotechnology, Saint-Petersburg State University, 199034 Saint Petersburg, Russia; [email protected] 3 International Laboratory of Computer Technologies, ITMO University, 197101 Saint Petersburg, Russia; [email protected] 4 Laboratory of Mutagenesis and Genetic Toxicology, Vavilov Institute of General Genetics, Saint-Petersburg Branch, Russian Academy of Sciences, 199034 Saint Petersburg, Russia * Correspondence: [email protected] Received: 30 September 2020; Accepted: 13 November 2020; Published: 24 November 2020 Simple Summary: The etiology of cancer is linked to the occurrence of mutations during the reduplication of genetic material. Mutations leading to low replication fidelity are the culprits of many hereditary and sporadic cancers. The archetype of the current model of replication fork was proposed 30 years ago. In the sequel to our 2010 review with the words “years after” in the title inspired by A. Dumas’s novels, we go over new developments in the DNA replication field and analyze how they help elucidate the effects of the genetic variants of DNA polymerases on cancer. Abstract: Recent studies on tumor genomes revealed that mutations in genes of replicative DNA polymerases cause a predisposition for cancer by increasing genome instability. The past 10 years have uncovered exciting details about the structure and function of replicative DNA polymerases and the replication fork organization. -

The Genetic Program of Pancreatic Beta-Cell Replication in Vivo
Page 1 of 65 Diabetes The genetic program of pancreatic beta-cell replication in vivo Agnes Klochendler1, Inbal Caspi2, Noa Corem1, Maya Moran3, Oriel Friedlich1, Sharona Elgavish4, Yuval Nevo4, Aharon Helman1, Benjamin Glaser5, Amir Eden3, Shalev Itzkovitz2, Yuval Dor1,* 1Department of Developmental Biology and Cancer Research, The Institute for Medical Research Israel-Canada, The Hebrew University-Hadassah Medical School, Jerusalem 91120, Israel 2Department of Molecular Cell Biology, Weizmann Institute of Science, Rehovot, Israel. 3Department of Cell and Developmental Biology, The Silberman Institute of Life Sciences, The Hebrew University of Jerusalem, Jerusalem 91904, Israel 4Info-CORE, Bioinformatics Unit of the I-CORE Computation Center, The Hebrew University and Hadassah, The Institute for Medical Research Israel- Canada, The Hebrew University-Hadassah Medical School, Jerusalem 91120, Israel 5Endocrinology and Metabolism Service, Department of Internal Medicine, Hadassah-Hebrew University Medical Center, Jerusalem 91120, Israel *Correspondence: [email protected] Running title: The genetic program of pancreatic β-cell replication 1 Diabetes Publish Ahead of Print, published online March 18, 2016 Diabetes Page 2 of 65 Abstract The molecular program underlying infrequent replication of pancreatic beta- cells remains largely inaccessible. Using transgenic mice expressing GFP in cycling cells we sorted live, replicating beta-cells and determined their transcriptome. Replicating beta-cells upregulate hundreds of proliferation- related genes, along with many novel putative cell cycle components. Strikingly, genes involved in beta-cell functions, namely glucose sensing and insulin secretion were repressed. Further studies using single molecule RNA in situ hybridization revealed that in fact, replicating beta-cells double the amount of RNA for most genes, but this upregulation excludes genes involved in beta-cell function. -

Identification of Proteins Involved in the Maintenance of Genome Stability
Identification of Proteins Involved in the Maintenance of Genome Stability by Edith Hang Yu Cheng A thesis submitted in conformity with the requirements for the degree of Doctor of Philosophy Department of Biochemistry University of Toronto ©Copyright by Edith Cheng2015 Identification of Proteins Involved in the Maintenance of Genome Stability Edith Cheng Doctor of Philosophy Department of Biochemistry University of Toronto 2015 Abstract Aberrant changes to the genome structure underlie numerous human diseases such as cancers. The functional characterization ofgenesand proteins that maintain chromosome stability will be important in understanding disease etiology and developing therapeutics. I took a multi-faceted approach to identify and characterize genes involved in the maintenance of genome stability. As biological pathways involved in genome maintenance are highly conserved in evolution, results from model organisms can greatly facilitate functional discovery in humans. In S. cerevisiae, I identified 47 essential gene depletions with elevated levels of spontaneous DNA damage foci and 92 depletions that caused elevated levels of chromosome rearrangements. Of these, a core subset of 15 DNA replication genes demonstrated both phenotypes when depleted. Analysis of rearrangement breakpoints revealed enrichment at yeast fragile sites, Ty retrotransposons, early origins of replication and replication termination sites. Together, thishighlighted the integral role of DNA replicationin genome maintenance. In light of my findings in S. cerevisiae, I identified a list of 153 human proteins that interact with the nascentDNA at replication forks, using a DNA pull down strategy (iPOND) in human cell lines. As a complementary approach for identifying human proteins involved in genome ii maintenance, I usedthe BioID techniqueto discernin vivo proteins proximal to the human BLM- TOP3A-RMI1-RMI2 genome stability complex, which has an emerging role in DNA replication progression. -

The DNA Polymerases of Drosophila Melanogaster
Fly ISSN: 1933-6934 (Print) 1933-6942 (Online) Journal homepage: https://www.tandfonline.com/loi/kfly20 The DNA polymerases of Drosophila melanogaster Steven J. Marygold, Helen Attrill, Elena Speretta, Kate Warner, Michele Magrane, Maria Berloco, Sue Cotterill, Mitch McVey, Yikang Rong & Masamitsu Yamaguchi To cite this article: Steven J. Marygold, Helen Attrill, Elena Speretta, Kate Warner, Michele Magrane, Maria Berloco, Sue Cotterill, Mitch McVey, Yikang Rong & Masamitsu Yamaguchi (2020): The DNA polymerases of Drosophilamelanogaster, Fly, DOI: 10.1080/19336934.2019.1710076 To link to this article: https://doi.org/10.1080/19336934.2019.1710076 © 2020 The Author(s). Published by Informa UK Limited, trading as Taylor & Francis Group. View supplementary material Published online: 14 Jan 2020. Submit your article to this journal Article views: 342 View related articles View Crossmark data Full Terms & Conditions of access and use can be found at https://www.tandfonline.com/action/journalInformation?journalCode=kfly20 FLY https://doi.org/10.1080/19336934.2019.1710076 REVIEW The DNA polymerases of Drosophila melanogaster Steven J. Marygold a, Helen Attrill a, Elena Speretta b, Kate Warner b, Michele Magrane b, Maria Berloco c, Sue Cotterill d, Mitch McVey e, Yikang Rong f, and Masamitsu Yamaguchig aFlyBase, Department of Physiology, Development and Neuroscience, University of Cambridge, Cambridge, UK; bUniProt, European Molecular Biology Laboratory, European Bioinformatics Institute (EMBL-EBI), Cambridgeshire, UK; cDipartimento di Biologia, -

Bioinformatics Analysis of Differentially Expressed Genes and Pathways in the Development of Cervical Cancer Baojie Wu* and Shuyi Xi
Wu and Xi BMC Cancer (2021) 21:733 https://doi.org/10.1186/s12885-021-08412-4 RESEARCH Open Access Bioinformatics analysis of differentially expressed genes and pathways in the development of cervical cancer Baojie Wu* and Shuyi Xi Abstract Background: This study aimed to explore and identify key genes and signaling pathways that contribute to the progression of cervical cancer to improve prognosis. Methods: Three gene expression profiles (GSE63514, GSE64217 and GSE138080) were screened and downloaded from the Gene Expression Omnibus database (GEO). Differentially expressed genes (DEGs) were screened using the GEO2R and Venn diagram tools. Then, Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses were performed. Gene set enrichment analysis (GSEA) was performed to analyze the three gene expression profiles. Moreover, a protein–protein interaction (PPI) network of the DEGs was constructed, and functional enrichment analysis was performed. On this basis, hub genes from critical PPI subnetworks were explored with Cytoscape software. The expression of these genes in tumors was verified, and survival analysis of potential prognostic genes from critical subnetworks was conducted. Functional annotation, multiple gene comparison and dimensionality reduction in candidate genes indicated the clinical significance of potential targets. Results: A total of 476 DEGs were screened: 253 upregulated genes and 223 downregulated genes. DEGs were enriched in 22 biological processes, 16 cellular components and 9 molecular functions in precancerous lesions and cervical cancer. DEGs were mainly enriched in 10 KEGG pathways. Through intersection analysis and data mining, 3 key KEGG pathways and related core genes were revealed by GSEA.