Consequences and Resolution of Transcription–Replication Conflicts
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Biochemical Characterization of DDX43 (HAGE) Helicase
Biochemical Characterization of DDX43 (HAGE) Helicase A Thesis Submitted to the College of Graduate Studies and Research In Fulfillment of the Requirements For the Degree of Master of Science In the Department of Biochemistry University of Saskatchewan Saskatoon By Tanu Talwar © Copyright Tanu Talwar, March, 2017. All rights reserved PERMISSION OF USE STATEMENT I hereby present this thesis in partial fulfilment of the requirements for a postgraduate degree from the University of Saskatchewan and agree that the Libraries of this University may make it freely available for inspection. I further agree that permission for copying of this thesis in any manner, either in whole or in part, for scholarly purposes may be granted by the professor or professors who supervised this thesis or, in their absence, by the Head of the Department or the Dean of the College in which my thesis work was done. It is understood that any copying or publication or use of this thesis or parts of it for any financial gain will not be allowed without my written permission. It is also understood that due recognition shall be given to me and to the University of Saskatchewan in any scholarly use which may be made of any material in my thesis. Requests for permission to copy or to make other use of material in this thesis in whole or part should be addressed to: Head of the Department of Biochemistry University of Saskatchewan 107 Wiggins Road Saskatoon, Saskatchewan, Canada S7N 5E5 i ABSTRACT DDX43, DEAD-box polypeptide 43, also known as HAGE (helicase antigen gene), is a member of the DEAD-box family of RNA helicases. -

Gene Therapy Glossary of Terms
GENE THERAPY GLOSSARY OF TERMS A • Phase 3: A phase of research to describe clinical trials • Allele: one of two or more alternative forms of a gene that that gather more information about a drug’s safety and arise by mutation and are found at the same place on a effectiveness by studying different populations and chromosome. different dosages and by using the drug in combination • Adeno-Associated Virus: A single stranded DNA virus that has with other drugs. These studies typically involve more not been found to cause disease in humans. This type of virus participants.7 is the most frequently used in gene therapy.1 • Phase 4: A phase of research to describe clinical trials • Adenovirus: A member of a family of viruses that can cause occurring after FDA has approved a drug for marketing. infections in the respiratory tract, eye, and gastrointestinal They include post market requirement and commitment tract. studies that are required of or agreed to by the study • Adeno-Associated Virus Vector: Adeno viruses used as sponsor. These trials gather additional information about a vehicles for genes, whose core genetic material has been drug’s safety, efficacy, or optimal use.8 removed and replaced by the FVIII- or FIX-gene • Codon: a sequence of three nucleotides in DNA or RNA • Amino Acids: building block of a protein that gives instructions to add a specific amino acid to an • Antibody: a protein produced by immune cells called B-cells elongating protein in response to a foreign molecule; acts by binding to the • CRISPR: a family of DNA sequences that can be cleaved by molecule and often making it inactive or targeting it for specific enzymes, and therefore serve as a guide to cut out destruction and insert genes. -

And Embryo-Expressed KHDC1/DPPA5/ECAT1/OOEP Gene Family
Available online at www.sciencedirect.com Genomics 90 (2007) 583–594 www.elsevier.com/locate/ygeno Atypical structure and phylogenomic evolution of the new eutherian oocyte- and embryo-expressed KHDC1/DPPA5/ECAT1/OOEP gene family Alice Pierre a, Mathieu Gautier b, Isabelle Callebaut c, Martine Bontoux a, Eric Jeanpierre a, ⁎ Pierre Pontarotti d, Philippe Monget a, a Physiologie de la Reproduction et des Comportements, UMR 6175 INRA–CNRS–Université F. Rabelais de Tours Haras Nationaux, 37380 Nouzilly, France b Laboratoire de Génétique Biochimique et de Cytogénétique, Domaine de Vilvert, INRA, 78352 Jouy-en-Josas, France c Département de Biologie Structurale, Institut de Minéralogie et de Physique des Milieux Condensés, UMR CNRS 7590, Université Pierre et Marie Curie–Paris 6, Université Denis Diderot–Paris 7, IPGP, 140 Rue de Lourmel, 75015 Paris, France d EA 3781 Evolution Biologique, Université d’Aix Marseille I, 13331 Marseille Cedex 3, France Received 12 April 2007; accepted 12 June 2007 Available online 3 October 2007 Abstract Several recent studies have shown that genes specifically expressed by the oocyte are subject to rapid evolution, in particular via gene duplication mechanisms. In the present work, we have focused our attention on a family of genes, specific to eutherian mammals, that are located in unstable genomic regions. We have identified two genes specifically expressed in the mouse oocyte: Khdc1a (KH homology domain containing 1a, also named Ndg1 for Nur 77 downstream gene 1, a target gene of the Nur77 orphan receptor), and another gene structurally related to Khdc1a that we have renamed Khdc1b. In this paper, we show that Khdc1a and Khdc1b belong to a family of several members including the so-called developmental pluripotency A5 (Dppa5) genes, the cat/dog oocyte expressed protein (cat OOEP and dog OOEP) genes, and the ES cell- associated transcript 1 (Ecat1) genes. -
Supplementary Text and Figures.Pdf
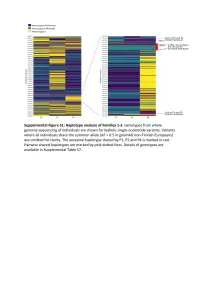
Supplemental Figure S2. PRIM1 immunoblots (20-75 kDa). (A) Corresponds to Figure 2D, with additional bands seen in P2 at 75 kDa and 25 kDa. (B) Independent experiment with cell lysates from C and P2 in which additional bands at 25 kDa not evident. (C) Corresponds to Figure 2D. No additional band visible at 25 kDa. (D) Validation of monoclonal antibody (8G10) with siRNA to PRIM1 in HeLa cells. siLUC, luciferase negative control. siPRIM1, siRNA targeting PRIM1 transcript. Vinculin used as a loading control. Supplemental Figure S3: Predicted destabilisation of PRIM1 by the C301R substitution. (A) Cysteine 301 lies in a buried hydrophobic region in PRIM1. DNA primase dimer crystal structure (PDB: 4BPU). PRIM1 residues shaded according to solvent accessibility. Cysteine 301, substituted to arginine in P5, lies in a buried hydrophobic region. (B) Cys301 is evolutionarily conserved in vertebrates. Neither arginine or other large or charged amino acids are observed at this position in other species. (C) The C301R substitution observed in P5 is predicted to lead to destabilisation of all available PRIM1 crystal structures. In contrast, substitution with leucine or threonine, as observed in some orthologous proteins, is not predicted to lead to destabilisation. Data points, predicted changes in free-energy (ΔΔG) from FoldX plotted for each of 14 available crystal structures. (D) Immunoblotting of RPE1 cells transfected with dual reporter constructs (as described in Figure 3B and Materials and Methods) confirms production of PRIM1-GFP and FLAG-SR at expected molecular weights and shows reduced protein levels for the C301R variant. n=2 experiments shown. C301R protein levels relative to WT after normalization to actin loading control indicated underneath the blot. -

A MUTYH Germline Mutation Is Associated with Small Intestinal
248 J P Dumanski et al. MUTYH substitution 24:8 427–443 Research Gly396Asp in SI-NETs Open Access A MUTYH germline mutation is associated with small intestinal neuroendocrine tumors Jan P Dumanski1,*, Chiara Rasi1,*, Peyman Björklund2, Hanna Davies1, Abir S Ali3, Malin Grönberg3, Staffan Welin3, Halfdan Sorbye4,5, Henning Grønbæk6, Janet L Cunningham7, Lars A Forsberg1, Lars Lind8, Erik Ingelsson9, Peter Stålberg10, Per Hellman10 and Eva Tiensuu Janson3 1Department of Immunology, Genetics and Pathology and SciLifeLab, Uppsala University, Uppsala, Sweden 2Department of Surgical Sciences, Experimental Surgery, Uppsala University, Uppsala, Sweden 3Department of Medical Sciences, Endocrine Oncology, Uppsala University, Uppsala, Sweden 4Department of Oncology, Haukeland University Hospital, Bergen, Norway 5Department of Clinical Science, University of Bergen, Bergen, Norway 6Department of Hepatology and Gastroenterology, Aarhus University Hospital, Aarhus, Denmark 7 Department of Neuroscience, Uppsala University, Uppsala, Sweden Correspondence 8 Department of Medical Sciences, Uppsala University, Uppsala, Sweden should be addressed 9 Division of Cardiovascular Medicine, Department of Medicine, Stanford University, San Francisco, California, USA to E Tiensuu Janson 10 Department of Surgical Sciences, Endocrine Surgery, Uppsala University, Uppsala, Sweden Email *(J P Dumanski and C Rasi shared first co-authorship) eva.tiensuu_janson@medsci. uu.se Abstract The genetics behind predisposition to small intestinal neuroendocrine tumors (SI-NETs) Key Words Endocrine-Related Cancer Endocrine-Related is largely unknown, but there is growing awareness of a familial form of the disease. f familial cancer We aimed to identify germline mutations involved in the carcinogenesis of SI-NETs. f cancer predisposition The strategy included next-generation sequencing of exome- and/or whole-genome of f small intestinal carcinoid blood DNA, and in selected cases, tumor DNA, from 24 patients from 15 families with f oxidative stress the history of SI-NETs. -

COMMENTARY Does Transcription by RNA Polymerase Play a Direct Role in the Initiation of Replication?
Journal of Cell Science 107, 1381-1387 (1994) 1381 Printed in Great Britain © The Company of Biologists Limited 1994 COMMENTARY Does transcription by RNA polymerase play a direct role in the initiation of replication? A. Bassim Hassan* and Peter R. Cook† CRC Nuclear Structure and Function Research Group, Sir William Dunn School of Pathology, University of Oxford, South Parks Road, Oxford, UK *Present address: Addenbrooke’s NHS Trust, Hills Rd, Cambridge CB2 2QQ, UK †Author for correspondence SUMMARY RNA polymerases have been implicated in the initiation of replication in bacteria. The conflicting evidence for a role in initiation in eukaryotes is reviewed. Key words: cell cycle, initiation, origin, replication, transcription PRIMERS AND PRIMASES complex becomes exclusively involved in the synthesis of Okazaki fragments on the lagging strand. A critical step in this DNA polymerases cannot initiate the synthesis of new DNA process is the unwinding of the duplex. chains, they can only elongate pre-existing primers. The opposite polarities of the two strands of the double helix coupled with the 5′r3′ polarity of the polymerase means that RNA POLYMERASES AND THE INITIATION OF replication occurs relatively continuously on one (leading) REPLICATION IN BACTERIA strand and discontinuously on the other (lagging) strand. The continuous strand probably needs to be primed once, usually The first evidence of a role for RNA polymerase came from at an origin. Nature has found many different ways of doing the demonstration that the initiation of replication in this, including the use of RNA primers made by an RNA poly- Escherichia coli was sensitive to rifampicin, an inhibitor of merase (e.g. -

Non-Coding Transcription Influences the Replication Initiation Program Through Chromatin Regulation
Downloaded from genome.cshlp.org on October 9, 2021 - Published by Cold Spring Harbor Laboratory Press Non-coding transcription influences the replication initiation program through chromatin regulation Julien Soudet1*, Jatinder Kaur Gill1 and Françoise Stutz1*. 1Dept. of Cell Biology, University of Geneva, Switzerland. *Correspondence: [email protected], [email protected] Keywords (10 words): non-coding RNA, non-coding transcription, histone modifications, nucleosomes, replication timing, replication initiation, yeast 1 Downloaded from genome.cshlp.org on October 9, 2021 - Published by Cold Spring Harbor Laboratory Press ABSTRACT In Eukaryotic organisms, replication initiation follows a temporal program. Among the parameters that regulate this program in Saccharomyces cerevisiae, chromatin structure has been at the center of attention without considering the contribution of transcription. Here, we revisit the replication initiation program in the light of widespread genomic non-coding transcription. We find that non-coding RNA transcription termination in the vicinity of ARS (Autonomously Replicating Sequences) shields replication initiation from transcriptional readthrough. Consistently, high natural nascent transcription correlates with low ARS efficiency and late replication timing. High readthrough transcription is also linked to increased nucleosome occupancy and high levels of H3K36me3. Moreover, forcing ARS readthrough transcription promotes these chromatin features. Finally, replication initiation defects induced by increased transcriptional readthrough are partially rescued in the absence of H3K36 methylation. Altogether, these observations indicate that natural non-coding transcription into ARS influences replication initiation through chromatin regulation. 2 Downloaded from genome.cshlp.org on October 9, 2021 - Published by Cold Spring Harbor Laboratory Press INTRODUCTION DNA replication is a fundamental process occurring in all living organisms and ensuring accurate duplication of the genome. -

Ch 7 -Brock Information Flow in Biological Systems
Systems Microbiology Monday Oct 2 - Ch 7 -Brock Information flow in biological systems •• DNADNA replicationreplication •• TranscriptionTranscription •• TranslationTranslation Central Dogma DNA Replication Transcription Images removed due to copyright restrictions. RNA Reverse Transcription Translation Protein Flow of information replication DNA → DNA transcription ↓ RNA translation ↓ protein 5' end ring numbering system for -P-O-C deoxyribose 5’ -C O O 4’ 1’ P O- O 3’ 2’ C ssDNA 3’ end HO In a nucleotide, e.g., adenosine monophosphate (AMP), the base is bonded to a ribose sugar, which has a phosphate in ester linkage to the 5' hydroxyl. NH NH NH2 2 2 adenine N N N N N N N N N N N N H −2 HO O3P O CH CH 5' 2 O 2 O 4' H H 1' H H ribose H 3' 2' H H H OH OH OH OH adenine adenosine adenosine monophosphate (AMP) Nucleic acids have a NH2 backbone of adenine N N alternating Pi & ribose moieties. N NH − N 2 Phosphodiester 5' end O cytosine − 5' O P O CH N linkages form as the 2 O 4' 1' O H H ribose 5' phosphate of one N O H 3' 2' H nucleotide forms an O OH − 5' ester link with the 3' O P O CH 2 O OH of the adjacent O H H ribose nucleotide. H 3' H O OH − O P O (etc) nucleic acid 3' end O H N H O N Guanine Cytosine N H N N N N O H N Backbone Backbone Hydrogen H bond H O H N CH3 N Thymine N H N N Adenine N N Hydrogen O bond Backbone Backbone Figure by MIT OCW. -

Gene Expression Profiling Analysis Contributes to Understanding the Association Between Non-Syndromic Cleft Lip and Palate, and Cancer
2110 MOLECULAR MEDICINE REPORTS 13: 2110-2116, 2016 Gene expression profiling analysis contributes to understanding the association between non-syndromic cleft lip and palate, and cancer HONGYI WANG, TAO QIU, JIE SHI, JIULONG LIANG, YANG WANG, LIANGLIANG QUAN, YU ZHANG, QIAN ZHANG and KAI TAO Department of Plastic Surgery, General Hospital of Shenyang Military Area Command, PLA, Shenyang, Liaoning 110016, P.R. China Received March 10, 2015; Accepted December 18, 2015 DOI: 10.3892/mmr.2016.4802 Abstract. The present study aimed to investigate the for NSCL/P were implicated predominantly in the TGF-β molecular mechanisms underlying non-syndromic cleft lip, signaling pathway, the cell cycle and in viral carcinogenesis. with or without cleft palate (NSCL/P), and the association The TP53, CDK1, SMAD3, PIK3R1 and CASP3 genes were between this disease and cancer. The GSE42589 data set found to be associated, not only with NSCL/P, but also with was downloaded from the Gene Expression Omnibus data- cancer. These results may contribute to a better understanding base, and contained seven dental pulp stem cell samples of the molecular mechanisms of NSCL/P. from children with NSCL/P in the exfoliation period, and six controls. Differentially expressed genes (DEGs) were Introduction screened using the RankProd method, and their potential functions were revealed by pathway enrichment analysis and Non-syndromic cleft lip, with or without cleft palate (NSCL/P) construction of a pathway interaction network. Subsequently, is one of the most common types of congenital defect and cancer genes were obtained from six cancer databases, and affects 3.4-22.9/10,000 individuals worldwide (1). -

PRIM1 Polyclonal ANTIBODY Catalog Number:10773-1-AP 2 Publications
For Research Use Only PRIM1 Polyclonal ANTIBODY www.ptglab.com Catalog Number:10773-1-AP 2 Publications Catalog Number: GenBank Accession Number: Purification Method: Basic Information 10773-1-AP BC005266 Antigen affinity purification Size: GeneID (NCBI): Recommended Dilutions: 150UL , Concentration: 133 μg/ml by 5557 WB 1:500-1:1000 Bradford method using BSA as the Full Name: IP 0.5-4.0 ug for IP and 1:200-1:1000 standard; primase, DNA, polypeptide 1 (49kDa) for WB Source: IHC 1:20-1:200 Calculated MW: IF 1:50-1:500 Rabbit 50 kDa Isotype: Observed MW: IgG 50 kDa Immunogen Catalog Number: AG1124 Applications Tested Applications: Positive Controls: IF, IHC, IP, WB, ELISA WB : K-562 cells, HeLa cells, Sp2/0 cells Cited Applications: IP : HeLa cells, IHC IHC : human lymphoma tissue, Species Specificity: human, mouse, rat IF : HeLa cells, Cited Species: mouse Note-IHC: suggested antigen retrieval with TE buffer pH 9.0; (*) Alternatively, antigen retrieval may be performed with citrate buffer pH 6.0 DNA replication in human cells is initiated by a complex apparatus containing a DNA polymerase-alpha/primase Background Information complex. Primase synthesizes oligoribonucleotides that serve as primers for the initiation of DNA synthesis. It plays a role in both the initiation of DNA replication and the synthesis of Okazaki fragments for lagging strand synthesis. PRIM1 is a subnuit of this complex. Notable Publications Author Pubmed ID Journal Application Pierre Kunz 30986223 PLoS One IHC Liam C Hunt 31365869 Cell Rep Storage: Storage Store at -20°C. Stable for one year after shipment. -

DNA : TACGCGTATACCGACATT Transcription Will Make Mrna From
Transcription and Translation Practice: Name _____________________________________ Background: • DNA controls our traits • DNA is found in the nucleus of our cells • Our traits are controlled by proteins • DNA is the instructions to make proteins • Proteins are made in ribosomes (outside the nucleus) • Proteins are made of amino acids Transcription makes RNA from DNA • RNA is complementary to DNA • RNA can leave the nucleus (DNA cannot) Translation makes proteins using RNA • Takes place at the ribosome • mRNA is “read” to put together a protein from amino acids Example: Beyonce has brown eyes. Her eyes look brown because her DNA codes for a brown pigment in the cells of her eyes. This is the gene that codes for brown eyes. DNA : T A C G C G T A T A C C G A C A T T Transcription will make mRNA from DNA mRNA: A U G C G C _________________________ Transcription will join amino acids to make the protein Rules for Transcription: Methionine - Arginine - ____________________________________ Base of DNA → Base in mRNA A → U Rules of Translation: C → G Three letters of mRNA = a codon G → C A codon “codes” for an amino acid We use the “Genetic Code” to determine the amino acids T → A RNA contains Uracil instead of Thymine The complete chain of amino acids will complete the protein that will give Beyonce her brown eyes. If you have brown eyes you have the same protein. If you have blue or green eyes your DNA sequence is a little different which will make the amino acid sequence (protein) a little different. -

Conversion of OX174 and Fd Single-Stranded DNA to Replicative Forms in Extracts of Escherichia Coli (Dnac, Dnad, and Dnag Gene Products/DNA Polymerase III) REED B
Proc. Nat. Acad. Sci. USA Vol. 69, No. 11, pp. 3233-3237, November 1972 Conversion of OX174 and fd Single-Stranded DNA to Replicative Forms in Extracts of Escherichia coli (dnaC, dnaD, and dnaG gene products/DNA polymerase III) REED B. WICKNER, MICHEL WRIGHT, SUE WICKNER, AND JERARD HURWITZ Department of Developmental Biology and Cancer, Division of Biological Sciences, Albert Einstein College of Medicine, Bronx, New York 10461 Communicated by Harry Eagle, August 28, 1972 ABSTRACT 4X174 and M13 (fd) single-stranded cir- MATERIALS AND METHODS cular DNAs are converted to their replicative forms by ex- tracts of E. coli pol Al cells. We find that the qX174 DNA- [a-32P]dTTP was obtained from New England Nuclear Corp. dependent reaction requires Mg++, ATP, and all four de- OX174 DNA was prepared by the method of Sinsheimer (7) oxynucleoside triphosphates, but not CTP, UTP, or GTP. or Franke and Ray (8), while fd viral DNA was prepared as This reaction also involves the products of the dnaC, dnaD, dnaE (DNA polymerase III), and dnaG genes, described (9). Pancreatic RNase was the highest grade ob- but not that of dnaF (ribonucleotide reductase). The in tainable from Worthington Biochemical Corp. It was further vitro conversion of fd single-stranded DNA to the replica- freed of possible contaminating DNase by heating a solution tive form requires all four ribonucleoside triphosphates, (2 mg/ml in 15 mM sodium citrate, pH 5) at 800 for 10 min. Mg++, and all four deoxynucleoside triphosphates. The E. protein was purified from E. coli strain reaction involves the product of gene dnaE but not those coli unwinding of genes dnaC, dnaD, dnaF, or dnaG.