Inferring Ancestry Across Hybrid Genomes Using Low-Coverage Sequence Data
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

THE CASE AGAINST Marine Mammals in Captivity Authors: Naomi A
s l a m m a y t T i M S N v I i A e G t A n i p E S r a A C a C E H n T M i THE CASE AGAINST Marine Mammals in Captivity The Humane Society of the United State s/ World Society for the Protection of Animals 2009 1 1 1 2 0 A M , n o t s o g B r o . 1 a 0 s 2 u - e a t i p s u S w , t e e r t S h t u o S 9 8 THE CASE AGAINST Marine Mammals in Captivity Authors: Naomi A. Rose, E.C.M. Parsons, and Richard Farinato, 4th edition Editors: Naomi A. Rose and Debra Firmani, 4th edition ©2009 The Humane Society of the United States and the World Society for the Protection of Animals. All rights reserved. ©2008 The HSUS. All rights reserved. Printed on recycled paper, acid free and elemental chlorine free, with soy-based ink. Cover: ©iStockphoto.com/Ying Ying Wong Overview n the debate over marine mammals in captivity, the of the natural environment. The truth is that marine mammals have evolved physically and behaviorally to survive these rigors. public display industry maintains that marine mammal For example, nearly every kind of marine mammal, from sea lion Iexhibits serve a valuable conservation function, people to dolphin, travels large distances daily in a search for food. In learn important information from seeing live animals, and captivity, natural feeding and foraging patterns are completely lost. -

Brown Bear (Ursus Arctos) John Schoen and Scott Gende Images by John Schoen
Brown Bear (Ursus arctos) John Schoen and Scott Gende images by John Schoen Two hundred years ago, brown (also known as grizzly) bears were abundant and widely distributed across western North America from the Mississippi River to the Pacific and from northern Mexico to the Arctic (Trevino and Jonkel 1986). Following settlement of the west, brown bear populations south of Canada declined significantly and now occupy only a fraction of their original range, where the brown bear has been listed as threatened since 1975 (Servheen 1989, 1990). Today, Alaska remains the last stronghold in North America for this adaptable, large omnivore (Miller and Schoen 1999) (Fig 1). Brown bears are indigenous to Southeastern Alaska (Southeast), and on the northern islands they occur in some of the highest-density FIG 1. Brown bears occur throughout much of southern populations on earth (Schoen and Beier 1990, Miller et coastal Alaska where they are closely associated with salmon spawning streams. Although brown bears and grizzly bears al. 1997). are the same species, northern and interior populations are The brown bear in Southeast is highly valued by commonly called grizzlies while southern coastal populations big game hunters, bear viewers, and general wildlife are referred to as brown bears. Because of the availability of abundant, high-quality food (e.g. salmon), brown bears enthusiasts. Hiking up a fish stream on the northern are generally much larger, occur at high densities, and have islands of Admiralty, Baranof, or Chichagof during late smaller home ranges than grizzly bears. summer reveals a network of deeply rutted bear trails winding through tunnels of devil’s club (Oplopanx (Klein 1965, MacDonald and Cook 1999) (Fig 2). -
Crystal Reports Activex Designer
Quiz List—Reading Practice Page 1 Printed Wednesday, March 18, 2009 2:36:33PM School: Churchland Academy Elementary School Reading Practice Quizzes Quiz Word Number Lang. Title Author IL ATOS BL Points Count F/NF 9318 EN Ice Is...Whee! Greene, Carol LG 0.3 0.5 59 F 9340 EN Snow Joe Greene, Carol LG 0.3 0.5 59 F 36573 EN Big Egg Coxe, Molly LG 0.4 0.5 99 F 9306 EN Bugs! McKissack, Patricia C. LG 0.4 0.5 69 F 86010 EN Cat Traps Coxe, Molly LG 0.4 0.5 95 F 9329 EN Oh No, Otis! Frankel, Julie LG 0.4 0.5 97 F 9333 EN Pet for Pat, A Snow, Pegeen LG 0.4 0.5 71 F 9334 EN Please, Wind? Greene, Carol LG 0.4 0.5 55 F 9336 EN Rain! Rain! Greene, Carol LG 0.4 0.5 63 F 9338 EN Shine, Sun! Greene, Carol LG 0.4 0.5 66 F 9353 EN Birthday Car, The Hillert, Margaret LG 0.5 0.5 171 F 9305 EN Bonk! Goes the Ball Stevens, Philippa LG 0.5 0.5 100 F 7255 EN Can You Play? Ziefert, Harriet LG 0.5 0.5 144 F 9314 EN Hi, Clouds Greene, Carol LG 0.5 0.5 58 F 9382 EN Little Runaway, The Hillert, Margaret LG 0.5 0.5 196 F 7282 EN Lucky Bear Phillips, Joan LG 0.5 0.5 150 F 31542 EN Mine's the Best Bonsall, Crosby LG 0.5 0.5 106 F 901618 EN Night Watch (SF Edition) Fear, Sharon LG 0.5 0.5 51 F 9349 EN Whisper Is Quiet, A Lunn, Carolyn LG 0.5 0.5 63 NF 74854 EN Cooking with the Cat Worth, Bonnie LG 0.6 0.5 135 F 42150 EN Don't Cut My Hair! Wilhelm, Hans LG 0.6 0.5 74 F 9018 EN Foot Book, The Seuss, Dr. -

Docent Manual
2018 Docent Manual Suzi Fontaine, Education Curator Montgomery Zoo and Mann Wildlife Learning Museum 7/24/2018 Table of Contents Docent Information ....................................................................................................................................................... 2 Dress Code................................................................................................................................................................. 9 Feeding and Cleaning Procedures ........................................................................................................................... 10 Docent Self-Evaluation ............................................................................................................................................ 16 Mission Statement .................................................................................................................................................. 21 Education Program Evaluation Form ...................................................................................................................... 22 Education Master Plan ............................................................................................................................................ 23 Animal Diets ............................................................................................................................................................ 25 Mammals .................................................................................................................................................................... -
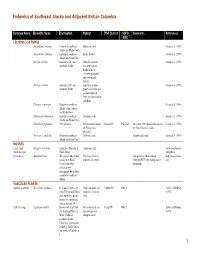
1 Endemics of Southeast Alaska and Adjacent British Columbia
Endemics of Southeast Alaska and Adjacent British Columbia Common Name Scientific Name Distribution Habitat TNF District ADFG Comments References GMU LICHENS and FUNGI Amygdalaria continua Endemic to southeast Subalpine rocks Geiser et al. (1998) Alaska and Haida Gwaii Amygdalaria haidensis Endemic to southeast Rocky habitats Geiser et al. (1998) Alaska and Haida Gwaii Bryoria carlottae Endemic to BC and Primarily on shore Geiser et al. (1998) southeast Alaska pine and western hemlock in low elevation peatlands and open mixed forests. Bryoria cervinula Endemic to BC and Primarily on open Geiser et al. (1998) southeast Alaska grown shore pine and western hemlock, from low elevations to subalpine. Placopsis roseonigra Endemic to southeast Geiser et al. (1998) Alaska (Sitka, Juneau) and Haida Gwaii Rhizocarpon hensseniae Endemic to southeast On alpine rocks Geiser et al. (1998) Alaska and Haida Gwaii Tremella hypogymniae NW of Haines Lichenicolous fungus Juneau RD GMU 1D Only other NA reports of this species Geiser et al. (1998) on Hypogymnia are from Ontario, Canada physodes. Verrucaria schofieldii Endemic to southeast On littoral rock. Recently described Geiser et al. (1998) Alaska and Haida Gwaii MOSSES Carey small Seligeria careyanna Endemic to Moresby I., Limestone cliffs www.forestbiodive limestone moss Haida Gwaii rsityinbc.ca a peat moss Sphagnum wilfii The type locality of this Pine bogs at low to This species is on the British http://www.efloras. species is in Haida moderate elevations Columbia RED list (endangered or org Gwaii. It has been threatened). collected only infrequently but is fairly common in southeast Alaska. VASCULAR PLANTS upswept moonwart Botrychium ascendens In Alaska, known only Mesic meadows and Yakutat RD GMU 5 Lipkin and Murray from Yakutat and Glacier sandy sites near sea (1997) Bay; elsewhere, from a level. -

The History of the Tall Ship Regina Maris
Linfield University DigitalCommons@Linfield Linfield Alumni Book Gallery Linfield Alumni Collections 2019 Dreamers before the Mast: The History of the Tall Ship Regina Maris John Kerr Follow this and additional works at: https://digitalcommons.linfield.edu/lca_alumni_books Part of the Cultural History Commons, and the United States History Commons Recommended Citation Kerr, John, "Dreamers before the Mast: The History of the Tall Ship Regina Maris" (2019). Linfield Alumni Book Gallery. 1. https://digitalcommons.linfield.edu/lca_alumni_books/1 This Book is protected by copyright and/or related rights. It is brought to you for free via open access, courtesy of DigitalCommons@Linfield, with permission from the rights-holder(s). Your use of this Book must comply with the Terms of Use for material posted in DigitalCommons@Linfield, or with other stated terms (such as a Creative Commons license) indicated in the record and/or on the work itself. For more information, or if you have questions about permitted uses, please contact [email protected]. Dreamers Before the Mast, The History of the Tall Ship Regina Maris By John Kerr Carol Lew Simons, Contributing Editor Cover photo by Shep Root Third Edition This work is licensed under the Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc- nd/4.0/. 1 PREFACE AND A TRIBUTE TO REGINA Steven Katona Somehow wood, steel, cable, rope, and scores of other inanimate materials and parts create a living thing when they are fastened together to make a ship. I have often wondered why ships have souls but cars, trucks, and skyscrapers don’t. -

Sing! 1975 – 2014 Song Index
Sing! 1975 – 2014 song index Song Title Composer/s Publication Year/s First line of song 24 Robbers Peter Butler 1993 Not last night but the night before ... 59th St. Bridge Song [Feelin' Groovy], The Paul Simon 1977, 1985 Slow down, you move too fast, you got to make the morning last … A Beautiful Morning Felix Cavaliere & Eddie Brigati 2010 It's a beautiful morning… A Canine Christmas Concerto Traditional/May Kay Beall 2009 On the first day of Christmas my true love gave to me… A Long Straight Line G Porter & T Curtan 2006 Jack put down his lister shears to join the welders and engineers A New Day is Dawning James Masden 2012 The first rays of sun touch the ocean, the golden rays of sun touch the sea. A Wallaby in My Garden Matthew Hindson 2007 There's a wallaby in my garden… A Whole New World (Aladdin's Theme) Words by Tim Rice & music by Alan Menken 2006 I can show you the world. A Wombat on a Surfboard Louise Perdana 2014 I was sitting on the beach one day when I saw a funny figure heading my way. A.E.I.O.U. Brian Fitzgerald, additional words by Lorraine Milne 1990 I can't make my mind up- I don't know what to do. Aba Daba Honeymoon Arthur Fields & Walter Donaldson 2000 "Aba daba ... -" said the chimpie to the monk. ABC Freddie Perren, Alphonso Mizell, Berry Gordy & Deke Richards 2003 You went to school to learn girl, things you never, never knew before. Abiyoyo Traditional Bantu 1994 Abiyoyo .. -

Large Inventoried Roadless Areas on the Tongass National Forest
Conservation Significance of Large Inventoried Roadless Areas on the Tongass National Forest North Prince of Wales Island Key to Symbols � Roads � Salmon streams Core Areas of Biological Value Past Clearcuts � Other streams Large Inventoried Roadless Areas M Old-growth Forests in Roadless Areas at Risk from Logging under the 2019 Alaska Roadless Rule DraftEIS, Preferred Alternative (Alt 6) December, 2019 David M. Albert CONSERVATION SIGNIFICANCE OF LARGE INVENTORIED ROADLESS AREAS ON THE TONGASS NATIONAL FOREST David M. Albert, Juneau AK ABSTRACT We evaluated the conservation significance of large inventoried roadless toward the goal of maintaining viable and well-distributed populations of fish and wildlife across the Tongass National Forest. We used the best available data to calculate indicators of habitat condition for 5 important species and forest systems. The significance of roadless areas was evaluated based the relative distribution of habitat values among biogeographic provinces, the degree to which habitats have been altered relative to historical conditions, the proportion of remaining values contained in large inventoried roadless areas; and the proportion of remaining values in lands potentially available for future development. No biological indicators exceeded the 40% threshold based on current alteration from original conditions region-wide, although loss of contiguous forest landscapes was approaching that value with a decline of 39.2%. However, within biogeographic provinces 25% of all indicators exceeded this threshold, with highest levels of alteration within the Prince of Wales Island group. The average decline across all indicators was 29% from historical conditions, regionwide. Consideration of lands potentially available for future development with removal of the Roadless Rule would result in a Cumulative Risk Index of 50.4% across all indicators. -

Wildlife Terrestrial Habitat Question 1 Appendix - MIS Management Indicator Species Monitoring Report
Wildlife Terrestrial Habitat Question 1 Appendix - MIS Management Indicator Species Monitoring Report This appendix reports on populations and habitat trends of Management Indicator Species (MIS) in accordance with the terrestrial wildlife goals of the Forest Plan. The Tongass Land and Resource Management Plan, monitoring question on Terrestrial Wildlife Habitat MIS states, “Are population and habitat trends for Management Indicator Species consistent with expectations? Are these trends due to changes in habitat conditions or other factors? If they are tied to habitat conditions, is there a direct relationship with forest management, climate change, or other factors? Terrestrial MIS include Red Squirrel, Black Bear, Brown Bear, Marten, River Otter, Sitka Black-tailed deer, Mountain Goat, Gray Wolf, Vancouver Canada Goose, Bald Eagle, Red-breasted Sapsucker, Hairy Woodpecker, and Brown Creeper.” In response to this monitoring question, the data relative to the MIS species is summarized and trends are analyzed species by species. The 1982 regulations to implement the National Forest Management Act require that Management Indicator Species (MIS) be identified as part of each forest plan. MIS serve multiple functions in forest planning: 1) MIS establish explicit Forest Plan objectives for fish and wildlife habitat, 2) MIS facilitate analysis of Forest Plan alternatives, and 3) MIS provide a means to monitor the effect of forest plan implementation. Much of the direction for MIS is outlined in CFR 219.19, together with direction for ecosystem diversity and species diversity. As such, MIS represents one part of a broader fabric of biodiversity and species management. A main criterion for the selection of the Tongass MIS was whether or not timber harvest activities potentially affect their key habitats. -

The Image and Cult of the Black Christ in Colonial Mexico and Central America
City University of New York (CUNY) CUNY Academic Works All Dissertations, Theses, and Capstone Projects Dissertations, Theses, and Capstone Projects 10-2014 Disseminating Devotion: The Image and Cult of the Black Christ in Colonial Mexico and Central America Elena FitzPatrick Sifford Graduate Center, City University of New York How does access to this work benefit ou?y Let us know! More information about this work at: https://academicworks.cuny.edu/gc_etds/332 Discover additional works at: https://academicworks.cuny.edu This work is made publicly available by the City University of New York (CUNY). Contact: [email protected] DISSEMINATING DEVOTION: THE IMAGE AND CULT OF THE BLACK CHRIST IN COLONIAL MEXICO AND CENTRAL AMERICA by ELENA FITZPATRICK SIFFORD A dissertation submitted to the Graduate Faculty in Art History in partial fulfillment of the requirements for the degree of Doctor of Philosophy, The City University of New York 2014 ©2014 ELENA FITZPATRICK SIFFORD All Rights Reserved ii This manuscript has been read and accepted for the Graduate Faculty in Art History in satisfaction of the dissertation requirement for the degree of Doctor of Philosophy. Date Dr. Eloise Quiñones Keber Chair of Examining Committee ____________________ ________________________________________ Date Dr. Claire Bishop Acting Executive Officer ____________________ ________________________________________ ___________________________________ Dr. Amanda Wunder ____________________________________ Dr. Timothy Pugh ____________________________________ Dr. Lauren Kilroy-Ewbank Supervisory Committee THE CITY UNIVERSITY OF NEW YORK iii Abstract DISSEMINATING DEVOTION: THE IMAGE AND CULT OF THE BLACK CHRIST IN COLONIAL MEXICO AND CENTRAL AMERICA by Elena FitzPatrick Sifford Adviser: Dr. Eloise Quiñones Keber Following the conquest of Mexico in 1521, Spanish conquerors and friars considered it their duty to bring Christianity to the New World. -

Genomic Evidence of Geographically Widespread Effect of Gene Flow From
Received Date : 07-Feb-2014 Revised Date : 15-Nov-2014 Accepted Date : 26-Nov-2014 Article type : Original Article Genomic evidence of geographically widespread effect of gene flow from polar bears into brown bears James A Cahill1, Ian Stirling5, Logan Kistler3, Rauf Salamzade2, Erik Ersmark4, Tara L Fulton1, Mathias Stiller1,6 , Richard E Green2, and Beth Shapiro1§ Article 1Dept. of Ecology and Evolutionary Biology, University of California, Santa Cruz, 95064, USA. 2Dept. of Biomolecular Engineering, University of California, Santa Cruz, 95064, USA. 3Departments of Anthropology and Biology, The Pennsylvania State University, 516 Carpenter Building, University Park PA 16802. 4 Department of Bioinformatics and Genetics, Swedish Museum of Natural History, SE-104 05 Stockholm, Sweden. 5 Wildlife Research Division, Department of Environment, Edmonton, Canada 6 Department of Evolutionary Genetics, MaX Planck Institute for Evolutionary Anthropology, Deutscher Platz 6, D-04103 Leipzig, Germany. §Corresponding author Corresponding author email address: [email protected] This article has been accepted for publication and undergone full peer review but has not been through the copyediting, typesetting, pagination and proofreading process, which may lead to differences between this version and the Version of Record. Please cite this article as doi: 10.1111/mec.13038 Accepted This article is protected by copyright. All rights reserved. Abstract Polar bears are an arctic, marine adapted species that is closely related to brown bears. Genome analyses have shown that polar bears are distinct and genetically homogeneous in comparison to brown bears. However, these analyses have also revealed a remarkable episode of polar bear gene flow into the population of brown bears that colonized the Admiralty, Baranof, and Chichagof Islands (ABC Islands) of Alaska. -

SMALL CETACEANS, BIG PROBLEMS a Global Review of the Impacts of Hunting on Small Whales, Dolphins and Porpoises
SMALL CETACEANS, BIG PROBLEMS A global review of the impacts of hunting on small whales, dolphins and porpoises A report by Sandra Altherr and Nicola Hodgins Edited by Sue Fisher, Kate O’Connell, D.J. Schubert, and Dave Tilford SMALL CETACEANS, BIG PROBLEMS A global review of the impacts of hunting on small whales, dolphins and porpoises A report by Sandra Altherr and Nicola Hodgins Edited by Sue Fisher, Kate O’Connell, D.J. Schubert, and Dave Tilford ACKNOWLEDGEMENTS The authors would like to thank the following people and institutions for providing helpful input, information and photos: Stefan Austermühle and his organisation Mundo Azul, Conservation India, The Dolphin Project, Astrid Fuchs, Dr. Lindsay Porter, Vivian Romano, and Koen Van Waerebeek. Special thanks also to Ava Rinehart and Alexandra Alberg of the Animal Welfare Institute for the design and layout of this report. GLOSSARY AAWP CMS IMARPE NMFS Abidjan Aquatic Wildlife Partnership Convention on the Conservation of Instituto Del Mar Del Peru National Marine Fisheries Service Migratory Species of Wild Animals ACCOBAMS IUU NOAA Agreement on the Conservation COSWIC Illegal, Unreported and Unregulated National Oceanic and Atmospheric of Cetaceans of the Black Sea, Commission on the Status of Fishing Administration Mediterranean Sea and Contiguous Endangered Wildlife in Canada Atlantic Area IUCN SC CPW International Union for Conservation Scientific Committee (International AQUATIC WILD MEAT Collaborative Partnership on of Nature Whaling Commission) The products derived from aquatic Sustainable Wildlife Management mammals and sea turtles that are IWC SMALL CETACEANS used for subsistence food and DFO International Whaling Commission Small and large cetaceans is traditional uses, including shells, Federal Department of Fisheries & neither a biological, nor political bones and organs, as well as bait for Oceans Ministry (Canada) JCNB classification, but has evolved Greenlandic-Canadian Joint fisheries becoming a widely used semantic DWC Commission for Narwhal and Beluga term.