Mechanism of Drug-Induced Qt Interval Prolongation
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The QT Interval: How Long Is Too Long?
heart matters The QT interval: How long is too long? By Natalie K. Cox, RN Staff Nurse • Coronary Care Unit • McLeod Regional Medical Center • Florence, S.C. What’s the QT interval and why’s it so the appearance that the QT interval is pro- important? In this article, I’ll answer these longed. However, a wide QRS complex questions plus show you how to measure represents depolarization, and LQTS is a dis- the QT interval and how to recognize the order of repolarization. Sometimes the end types of long QT syndrome (LQTS) and of a T wave isn’t clearly defi ned, which can their symptoms, causes, and treatments. make it diffi cult to get the QT measurement. Irregular rhythms also make it diffi cult to The long and short of it obtain a consistent QT interval measure- The QT interval on the ECG is measured ment. For example, atrial fi brillation makes from the beginning of the QRS complex to it extremely diffi cult to measure a QT inter- the end of the T wave (see ECG components). val because fi nding a T wave isn’t always It represents the time it takes for the ventricles of the heart to depolarize and repolarize, or to ECG components contract and relax. The QT interval is longer when First positive deflection after the the heart rate is slower and short- ECG components P or Q wave er when the heart rate is faster. So it’s necessary to calculate the corrected QT interval (QTc) using the Bazett formula: QT interval divided by the square root of the R-R interval. -

The Postmortem Distribution of Vardenafil (Levitra | in an Aviation
Journal of Analytical Toxicology, Vol. 31, July/August 2007 The Postmortem Distribution of Vardenafil (Levitra| in an Aviation Accident Victim with an Unusually High Blood Concentration* Robert D. Johnson ~, Russell J. Lewis, and Mike K. Angler Downloaded from https://academic.oup.com/jat/article/31/6/328/682815 by guest on 27 September 2021 Civil Aerospace Medical Institute, Federal Aviation Administration, Analytical Toxicology and Accident Research Laboratory, AAM-610, CAMI Building, 6500 S. MacArthur Blvd., Oklahoma City, Oklahoma 73169-6901 I Abstract phodiesterase type 5 enzyme (PDE5) found predominantly in the penile corpus cavernosum (2-7). Vardenafil (tevitra) is one of the most widely prescribed Vardenafil undergoes hepatic metabolism, producing the treatments for erectile dysfunction. This report presents a active desethyl metabolite M1. M1 contributes to the ob- rapid and reliable method for the identification and quantification served pharmacological effects provided by vardenafil, as M1 of vardenafil in postmortem fluids and tissues, applies this method exhibits approximately 30% of the potency of the parent to a postmortem case, and describes the distribution of vardenafil in various fluids and tissues.This procedure utilizes sildenafil-d8, drug (1). Under steady-state conditions, the plasma concen- which is structurally closely related to vardenafil, as an tration of M1 is approximately 26% of that seen for vardenafil internal standard for more accurate and reliable quantitation. (1). After oral administration of vardenafil, peak plasma con- The method incorporates solid-phaseextraction and liquid centrations are obtained within 30-60 min (1). Vardenafil chromatography-tandem mass spectrometry (MS) and and its active metabolite have a terminal half-life of approx- MS-MS-MS utilizing an atmospheric pressure chemical imately 4-5 h (1). -
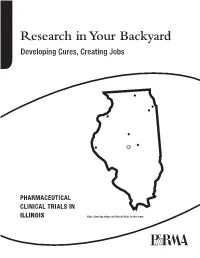
Research in Your Backyard Developing Cures, Creating Jobs
Research in Your Backyard Developing Cures, Creating Jobs PHARMACEUTICAL CLINICAL TRIALS IN ILLINOIS Dots show locations of clinical trials in the state. Executive Summary This report shows that biopharmaceutical research com- Quite often, biopharmaceutical companies hire local panies continue to be vitally important to the economy research institutions to conduct the tests and in Illinois, and patient health in Illinois, despite the recession. they help to bolster local economies in communities all over the state, including Chicago, Decatur, Joliet, Peoria, At a time when the state still faces significant economic Quincy, Rock Island, Rockford and Springfield. challenges, biopharmaceutical research companies are conducting or have conducted more than 4,300 clinical For patients, the trials offer another potential therapeutic trials of new medicines in collaboration with the state’s option. Clinical tests may provide a new avenue of care for clinical research centers, university medical schools and some chronic disease sufferers who are still searching for hospitals. Of the more than 4,300 clinical trials, 2,334 the medicines that are best for them. More than 470 of the target or have targeted the nation’s six most debilitating trials underway in Illinois are still recruiting patients. chronic diseases—asthma, cancer, diabetes, heart dis- ease, mental illnesses and stroke. Participants in clinical trials can: What are Clinical Trials? • Play an active role in their health care. • Gain access to new research treatments before they In the development of new medicines, clinical trials are are widely available. conducted to prove therapeutic safety and effectiveness and compile the evidence needed for the Food and Drug • Obtain expert medical care at leading health care Administration to approve treatments. -

Long QT Syndrome: from Channels to Cardiac Arrhythmias
Long QT syndrome: from channels to cardiac arrhythmias Arthur J. Moss, Robert S. Kass J Clin Invest. 2005;115(8):2018-2024. https://doi.org/10.1172/JCI25537. Review Series Long QT syndrome, a rare genetic disorder associated with life-threatening arrhythmias, has provided a wealth of information about fundamental mechanisms underlying human cardiac electrophysiology that has come about because of truly collaborative interactions between clinical and basic scientists. Our understanding of the mechanisms that control the critical plateau and repolarization phases of the human ventricular action potential has been raised to new levels through these studies, which have clarified the manner in which both potassium and sodium channels regulate this critical period of electrical activity. Find the latest version: https://jci.me/25537/pdf Review series Long QT syndrome: from channels to cardiac arrhythmias Arthur J. Moss1 and Robert S. Kass2 1Heart Research Follow-up Program, Department of Medicine, University of Rochester School of Medicine and Dentistry, Rochester, New York, USA. 2Department of Pharmacology, Columbia University Medical Center, New York, New York, USA. Long QT syndrome, a rare genetic disorder associated with life-threatening arrhythmias, has provided a wealth of information about fundamental mechanisms underlying human cardiac electrophysiology that has come about because of truly collaborative interactions between clinical and basic scientists. Our understanding of the mecha- nisms that control the critical plateau and repolarization phases of the human ventricular action potential has been raised to new levels through these studies, which have clarified the manner in which both potassium and sodium channels regulate this critical period of electrical activity. -

Anemia and the QT Interval in Hypertensive Patients
2084 International Journal of Collaborative Research on Internal Medicine & Public Health Anemia and the QT interval in hypertensive patients Ioana Mozos 1*, Corina Serban 2, Rodica Mihaescu 3 1 Department of Functional Sciences, “Victor Babes” University of Medicine and Pharmacy, Timisoara, Romania 2 Department of Functional Sciences, “Victor Babes” University of Medicine and Pharmacy, Timisoara, Romania 3 1st Department of Internal Medicine, “Victor Babes” University of Medicine and Pharmacy, Timisoara, Romania * Corresponding Author ; Email: [email protected] Abstract Introduction: A prolonged ECG QT interval duration and an increased QT dispersion (QTd) are predictors of sudden cardiac death. Anemia is known as a marker of adverse outcome in cardiovascular disease. Objective: The aim was to assess the relationship between anemia and QT intervals in hypertensive patients. Method: A total of 72 hypertensive patients underwent standard 12-lead ECG. QT intervals and QT dispersions were manually measured. Complete blood count was also assessed. Result: Linear regression analysis revealed significant associations between prolonged QTc and increased QTd and anemia and macrocytosis, respectively. Multiple regression analysis revealed a significant association between red cell distribution width (RDW) >15% and prolonged heart rate corrected maximal QT interval duration (QTc) and QT interval in lead DII (QTIIc). The most sensitive and specific predictor of prolonged QTc and QTIIc was anisocytosis. Anemia was the most sensitive predictor of -

Vardenafil Better Choice for Premature Ejaculation
August 15, 2005 • www.familypracticenews.com Men’s Health 47 Vardenafil Better Choice for Premature Ejaculation BY ROBERT FINN (24%) of the men and was secondary (in On a self-rating scale of 0-8, where 0 Center but is now at the University of San Francisco Bureau most cases to erectile dysfunction) in the means PE almost never, 4 means PE about Hamburg. remaining 26 men (77%). half the time, and 8 means PE almost al- Self-ratings of sexual satisfaction, on a 0- S AN A NTONIO — Vardenafil improved After a 4-week run-in period, 17 men ways, the mean score was 6.14 at baseline, 5 scale, where 0 means not at all satisfied premature ejaculation more than sertra- were given 10-mg vardenafil 10 minutes 4.28 with sertraline, and 3.2 with varde- and 5 means extremely satisfied, averaged line, Frank Sommer, M.D., reported at the before intercourse for 6 weeks. The other nafil. 1.4 at baseline, 3.2 with sertraline, and 4.2 annual meeting of the American Urolog- 17 received 50 mg of sertraline 4 hours be- IVELT, as measured by a stopwatch, with vardenafil. In addition, the partners’ ical Association. fore intercourse. averaged 0.54 minutes at baseline, 2.87 sexual satisfaction showed significant in- Both vardenafil (Levitra), a phosphodi- After a 1-week washout period, the men minutes with sertraline, and 5.23 minutes creases for sertraline and even more so for esterase-5 inhibitor, and sertraline (Zoloft), who had been receiving sertraline with vardenafil, reported Dr. Sommer, vardenafil. -

LEVITRA (Vardenafil Hcl) Tablets
LEVITRA (vardenafil HCl) Tablets DESCRIPTION LEVITRA® is an oral therapy for the treatment of erectile dysfunction. This monohydrochloride salt of vardenafil is a selective inhibitor of cyclic guanosine monophosphate (cGMP)-specific phosphodiesterase type 5 (PDE5). Vardenafil HCl is designated chemically as piperazine, 1-[[3-(1,4-dihydro-5- methyl-4-oxo-7-propylimidazo[5,1-f][1,2,4]triazin-2-yl)-4- ethoxyphenyl]sulfonyl]-4-ethyl-, monohydrochloride and has the following structural formula: O O HN N x HCl x 3H2O N N O S O N N Vardenafil HCl is a nearly colorless, solid substance with a molecular weight of 579.1 g/mol and a solubility of 0.11 mg/mL in water. LEVITRA is formulated as orange, round, film-coated tablets with "BAYER" cross debossed on one side and "2.5", "5", "10", and "20" on the other side corresponding to 2.5 mg, 5 mg, 10 mg, and 20 mg of vardenafil, respectively. In addition to the active ingredient, vardenafil HCl, each tablet contains microcrystalline cellulose, crospovidone, colloidal silicon dioxide, magnesium stearate, hypromellose, polyethylene glycol, titanium dioxide, yellow ferric oxide, and red ferric oxide. CLINICAL PHARMACOLOGY Mechanism of Action Penile erection is a hemodynamic process initiated by the relaxation of smooth muscle in the corpus cavernosum and its associated arterioles. During sexual stimulation, nitric oxide is released from nerve endings and endothelial cells in the corpus cavernosum. Nitric oxide activates the enzyme guanylate cyclase resulting in increased synthesis of cyclic guanosine monophosphate (cGMP) in the smooth muscle cells of the corpus cavernosum. The cGMP in turn triggers smooth muscle relaxation, allowing increased blood flow into the penis, resulting in erection. -

Phosphodiesterase (PDE)
Phosphodiesterase (PDE) Phosphodiesterase (PDE) is any enzyme that breaks a phosphodiester bond. Usually, people speaking of phosphodiesterase are referring to cyclic nucleotide phosphodiesterases, which have great clinical significance and are described below. However, there are many other families of phosphodiesterases, including phospholipases C and D, autotaxin, sphingomyelin phosphodiesterase, DNases, RNases, and restriction endonucleases, as well as numerous less-well-characterized small-molecule phosphodiesterases. The cyclic nucleotide phosphodiesterases comprise a group of enzymes that degrade the phosphodiester bond in the second messenger molecules cAMP and cGMP. They regulate the localization, duration, and amplitude of cyclic nucleotide signaling within subcellular domains. PDEs are therefore important regulators ofsignal transduction mediated by these second messenger molecules. www.MedChemExpress.com 1 Phosphodiesterase (PDE) Inhibitors, Activators & Modulators (+)-Medioresinol Di-O-β-D-glucopyranoside (R)-(-)-Rolipram Cat. No.: HY-N8209 ((R)-Rolipram; (-)-Rolipram) Cat. No.: HY-16900A (+)-Medioresinol Di-O-β-D-glucopyranoside is a (R)-(-)-Rolipram is the R-enantiomer of Rolipram. lignan glucoside with strong inhibitory activity Rolipram is a selective inhibitor of of 3', 5'-cyclic monophosphate (cyclic AMP) phosphodiesterases PDE4 with IC50 of 3 nM, 130 nM phosphodiesterase. and 240 nM for PDE4A, PDE4B, and PDE4D, respectively. Purity: >98% Purity: 99.91% Clinical Data: No Development Reported Clinical Data: No Development Reported Size: 1 mg, 5 mg Size: 10 mM × 1 mL, 10 mg, 50 mg (R)-DNMDP (S)-(+)-Rolipram Cat. No.: HY-122751 ((+)-Rolipram; (S)-Rolipram) Cat. No.: HY-B0392 (R)-DNMDP is a potent and selective cancer cell (S)-(+)-Rolipram ((+)-Rolipram) is a cyclic cytotoxic agent. (R)-DNMDP, the R-form of DNMDP, AMP(cAMP)-specific phosphodiesterase (PDE) binds PDE3A directly. -

Young Adults. Look for ST Elevation, Tall QRS Voltage, "Fishhook" Deformity at the J Point, and Prominent T Waves
EKG Abnormalities I. Early repolarization abnormality: A. A normal variant. Early repolarization is most often seen in healthy young adults. Look for ST elevation, tall QRS voltage, "fishhook" deformity at the J point, and prominent T waves. ST segment elevation is maximal in leads with tallest R waves. Note high take off of the ST segment in leads V4-6; the ST elevation in V2-3 is generally seen in most normal ECG's; the ST elevation in V2- 6 is concave upwards, another characteristic of this normal variant. Characteristics’ of early repolarization • notching or slurring of the terminal portion of the QRS wave • symmetric concordant T waves of large amplitude • relative temporal stability • most commonly presents in the precordial leads but often associated with it is less pronounced ST segment elevation in the limb leads To differentiate from anterior MI • the initial part of the ST segment is usually flat or convex upward in AMI • reciprocal ST depression may be present in AMI but not in early repolarization • ST segments in early repolarization are usually <2 mm (but have been reported up to 4 mm) To differentiate from pericarditis • the ST changes are more widespread in pericarditis • the T wave is normal in pericarditis • the ratio of the degree of ST elevation (measured using the PR segment as the baseline) to the height of the T wave is greater than 0.25 in V6 in pericarditis. 1 II. Acute Pericarditis: Stage 1 Pericarditis Changes A. Timing 1. Onset: Day 2-3 2. Duration: Up to 2 weeks B. Findings 1. -

Association of Oral Anticoagulants and Proton Pump Inhibitor Cotherapy with Hospitalization for Upper Gastrointestinal Tract Bleeding
Supplementary Online Content Ray WA, Chung CP, Murray KT, et al. Association of oral anticoagulants and proton pump inhibitor cotherapy with hospitalization for upper gastrointestinal tract bleeding. JAMA. doi:10.1001/jama.2018.17242 eAppendix. PPI Co-therapy and Anticoagulant-Related UGI Bleeds This supplementary material has been provided by the authors to give readers additional information about their work. Downloaded From: https://jamanetwork.com/ on 10/02/2021 Appendix: PPI Co-therapy and Anticoagulant-Related UGI Bleeds Table 1A Exclusions: end-stage renal disease Diagnosis or procedure code for dialysis or end-stage renal disease outside of the hospital 28521 – anemia in ckd 5855 – Stage V , ckd 5856 – end stage renal disease V451 – Renal dialysis status V560 – Extracorporeal dialysis V561 – fitting & adjustment of extracorporeal dialysis catheter 99673 – complications due to renal dialysis CPT-4 Procedure Codes 36825 arteriovenous fistula autogenous gr 36830 creation of arteriovenous fistula; 36831 thrombectomy, arteriovenous fistula without revision, autogenous or 36832 revision of an arteriovenous fistula, with or without thrombectomy, 36833 revision, arteriovenous fistula; with thrombectomy, autogenous or nonaut 36834 plastic repair of arteriovenous aneurysm (separate procedure) 36835 insertion of thomas shunt 36838 distal revascularization & interval ligation, upper extremity 36840 insertion mandril 36845 anastomosis mandril 36860 cannula declotting; 36861 cannula declotting; 36870 thrombectomy, percutaneous, arteriovenous -

The Relation Between Arterial Blood Pressure Variables and Ventricular Repolarization Parameters
860 International Journal of Collaborative Research on Internal Medicine & Public Health The relation between arterial blood pressure variables and ventricular repolarization parameters Ioana Mozos 1*, Corina Serban 2, Rodica Mihaescu 3 1 Department of Functional Sciences, “Victor Babes” University of Medicine and Pharmacy, Timisoara, Romania, [email protected] 2 Department of Functional Sciences, “Victor Babes” University of Medicine and Pharmacy, Timisoara, Romania 3 Department of Medical Semiology, “Victor Babes” University of Medicine and Pharmacy Timisoara, Romania ABSTRACT Introduction: Ventricular arrhythmia and sudden cardiac death risk are associated with prolonged electrocardiographic (ECG) QT and Tpeak-Tend intervals. Objective: To evaluate the influence of blood pressure variables on ventricular repolarization parameters, especially QT and Tpeak-Tend intervals. Method: Two groups of patients were enrolled in the study. The firs group included 77 patients, with essential hypertension, aged 62±12 years, 40% males. The control group included 56 patients, age and sex matched, with optimal, normal and high normal blood pressure. They underwent 12-lead ECG and ventricular repolarization parameters were assessed. QT intervals: QTmax (maximal QT interval duration), QTc (heart rate corrected QTmax), QTm (mean QT interval duration in all leads), QTIIc (heart rate corrected QT interval duration in lead DII), and T wave variables: T0e (maximal T wave duration), Tpe (maximal Tpeak-Tend interval) and Ta (maximal T wave amplitude) were manually measured. Arterial blood pressure variables: systolic (SBP), diastolic (DBP), mean arterial (MAP) and pulse pressure (PP), were recorded. Result: SBP was 139±24 mmHg, DBP 86±13 mmHg, MAP 103±15 mmHg, PP 53±16 mmHg, QTmax 430±51 ms, QTc 474±48 ms and Tpe 100±26 ms in the hypertensive group. -

Use of Sildenafil in Patients with Cardiovascular Disease
Arq Bras Cardiol GuimarãesReview et al volume 73, (nº6), 1999 Sildenafil in patients with cardiovascular disease Use of Sildenafil in Patients with Cardiovascular Disease Armênio Costa Guimarães, Marcus Vinícius Bolívar Malachias, Otávio Rizzi Coelho, Emílio Cesar Zilli, Rafael Leite Luna Introduction of phosphodiesterase inhibitors. The erectile action of sildenafil combines increase in arterial flow with reduction Erectile dysfunction, formerly called impotence, is the in the venous flow of cavernous body of penis. Sildenafil inability of the male to achieve or maintain penile erection and leads to relaxation of smooth muscle of penile arteries and thus engage in coitus1. It is common among patients with trabeculae surrounding the sinusoidal spaces, resulting in cardiovascular diseases or their risk factors. This dysfunc- a greater engorgement of cavernous body. The trabeculae tion occurs mainly among individuals with coronary artery of engorged sinusoidal spaces compress the penile disease, after episodes of acute ischemic syndrome, hyper- venules against the tunica albuginea, reducing venous tensive patients underpharmacologic treatment, and among flow, contributing to maintenance of engorgement of patients with heart failure. In approximately 85% of these ca- cavernous body8. Relaxation of this smooth muscle ses, the fear of a cardiac event during coitus constitutes an results from a decrease in intracellular calcium mediated important factor for erectile dysfunction 2-4. by accumulation of the second messenger, the cyclic Discovery of sildenafil citrate has represented a great de- guanosine monophosphate (cGMP), whose production velopment in the treatment of erectile dysfunction; it may results from activation of guanyl cyclase by nitric oxide benefit, among many others, those patients with cardiovascu- produced by the stimulus of endothelial cells generated lar diseases or with their risk factors 5.