Dissertation
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Role of RNA Editing in Cancer Development and Metabolic Disorders
Washington University School of Medicine Digital Commons@Becker Open Access Publications 2018 The oler of RNA editing in cancer development and metabolic disorders Che-Pei Kung Washington University School of Medicine in St. Louis Leonard B. Maggi Jr. Washington University School of Medicine in St. Louis Jason D. Weber Washington University School of Medicine in St. Louis Follow this and additional works at: https://digitalcommons.wustl.edu/open_access_pubs Recommended Citation Kung, Che-Pei; Maggi, Leonard B. Jr.; and Weber, Jason D., ,"The or le of RNA editing in cancer development and metabolic disorders." Frontiers in endocrinology.9,. 762. (2018). https://digitalcommons.wustl.edu/open_access_pubs/7400 This Open Access Publication is brought to you for free and open access by Digital Commons@Becker. It has been accepted for inclusion in Open Access Publications by an authorized administrator of Digital Commons@Becker. For more information, please contact [email protected]. REVIEW published: 18 December 2018 doi: 10.3389/fendo.2018.00762 The Role of RNA Editing in Cancer Development and Metabolic Disorders Che-Pei Kung 1,2*, Leonard B. Maggi Jr. 1,2 and Jason D. Weber 1,2,3* 1 ICCE Institute, Washington University School of Medicine, Saint Louis, MO, United States, 2 Division of Molecular Oncology, Department of Medicine, Washington University School of Medicine, Saint Louis, MO, United States, 3 Siteman Cancer Center, Department of Cell Biology and Physiology, Washington University School of Medicine, Saint Louis, MO, United States Numerous human diseases arise from alterations of genetic information, most notably DNA mutations. Thought to be merely the intermediate between DNA and protein, changes in RNA sequence were an afterthought until the discovery of RNA editing 30 years ago. -

Mohammad Karbaschi Thesis
STRUCTURAL, PHYSIOLOGICAL AND MOLECULAR CHARACTERISATION OF THE AUSTRALIAN NATIVE RESURRECTION GRASS TRIPOGON LOLIIFORMIS (F.MUELL.) C.E.HUBB. DURING DEHYDRATION AND REHYDRATION Mohammad Reza Karbaschi Submitted in fulfilment of the requirements for the degree of Doctor of Philosophy Centre for Tropical Crops and Biocommodities Science and Engineering Faculty Queensland University of Technology November 2015 Keywords Arabidopsis thaliana; Agrobacterium-mediated transformation; Anatomy; Anti-apoptotic proteins; BAG4; Escherichia coli; Bulliform cells; C4 photosynthesis; Cell wall folding; Cell membrane integrity; Chaperone-mediated autophagy; Chlorophyll fluorescence; Hsc70/Hsp70; Desiccation tolerance, Dehydration; Drought; Electrolyte leakage; Freehand sectioning; Homoiochlorophyllous; Leaf structure; Leaf folding; Reactive oxygen species (ROS); Resurrection plant; Morphology; Monocotyledon; Nicotiana benthamiana; Photosynthesis; Physiology; Plant tissue; Programed cell death (PCD); Propidium iodide staining; Protein microarray chip; Sclerenchymatous tissue; Stress; Structure; Tripogon loliiformis; Ubiquitin; Vacuole fragmentation; Kranz anatomy; XyMS+; Structural, physiological and molecular characterisation of the Australian native resurrection grass Tripogon loliiformis (F.Muell.) C.E.Hubb. during dehydration and rehydration i Abstract Plants, as sessile organisms must continually adapt to environmental changes. Water deficit is one of the major environmental stresses that affects plants. While most plants can tolerate moderate dehydration -
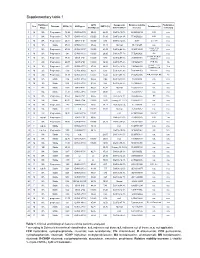
Supplementary Table 2 Supplementary Table 1
Supplementary table 1 Rai/ Binet IGHV Cytogenetic Relative viability Fludarabine- Sex Outcome CD38 (%) IGHV gene ZAP70 (%) Treatment (s) Stage identity (%) abnormalities* increase refractory 1 M 0/A Progressive 14,90 IGHV3-64*05 99,65 28,20 Del17p 18.0% 62,58322819 FCR n.a. 2 F 0/A Progressive 78,77 IGHV3-48*03 100,00 51,90 Del17p 24.8% 77,88052021 FCR n.a. 3 M 0/A Progressive 29,81 IGHV4-b*01 100,00 9,10 Del17p 12.0% 36,48 Len, Chl n.a. 4 M 1/A Stable 97,04 IGHV3-21*01 97,22 18,11 Normal 85,4191657 n.a. n.a. Chl+O, PCR, 5 F 0/A Progressive 87,00 IGHV4-39*07 100,00 43,20 Del13q 68.3% 35,23314039 n.a. HDMP+R 6 M 0/A Progressive 1,81 IGHV3-43*01 100,00 20,90 Del13q 77.7% 57,52490626 Chl n.a. Chl, FR, R-CHOP, 7 M 0/A Progressive 97,80 IGHV1-3*01 100,00 9,80 Del17p 88.5% 48,57389901 n.a. HDMP+R 8 F 2/B Progressive 69,07 IGHV5-a*03 100,00 16,50 Del17p 77.2% 107,9656878 FCR, BA No R-CHOP, FCR, 9 M 1/A Progressive 2,13 IGHV3-23*01 97,22 29,80 Del11q 16.3% 134,5866919 Yes Flavopiridol, BA 10 M 2/A Progressive 0,36 IGHV3-30*02 92,01 0,38 Del13q 81.9% 78,91844953 Unknown n.a. 11 M 2/B Progressive 15,17 IGHV3-20*01 100,00 13,20 Del11q 95.3% 75,52880995 FCR, R-CHOP, BR No 12 M 0/A Stable 0,14 IGHV3-30*02 90,62 7,40 Del13q 13.0% 13,0939004 n.a. -

Senescence Inhibits the Chaperone Response to Thermal Stress
SUPPLEMENTAL INFORMATION Senescence inhibits the chaperone response to thermal stress Jack Llewellyn1, 2, Venkatesh Mallikarjun1, 2, 3, Ellen Appleton1, 2, Maria Osipova1, 2, Hamish TJ Gilbert1, 2, Stephen M Richardson2, Simon J Hubbard4, 5 and Joe Swift1, 2, 5 (1) Wellcome Centre for Cell-Matrix Research, Oxford Road, Manchester, M13 9PT, UK. (2) Division of Cell Matrix Biology and Regenerative Medicine, School of Biological Sciences, Faculty of Biology, Medicine and Health, Manchester Academic Health Science Centre, University of Manchester, Manchester, M13 9PL, UK. (3) Current address: Department of Biomedical Engineering, University of Virginia, Box 800759, Health System, Charlottesville, VA, 22903, USA. (4) Division of Evolution and Genomic Sciences, School of Biological Sciences, Faculty of Biology, Medicine and Health, Manchester Academic Health Science Centre, University of Manchester, Manchester, M13 9PL, UK. (5) Correspondence to SJH ([email protected]) or JS ([email protected]). Page 1 of 11 Supplemental Information: Llewellyn et al. Chaperone stress response in senescence CONTENTS Supplemental figures S1 – S5 … … … … … … … … 3 Supplemental table S6 … … … … … … … … 10 Supplemental references … … … … … … … … 11 Page 2 of 11 Supplemental Information: Llewellyn et al. Chaperone stress response in senescence SUPPLEMENTAL FIGURES Figure S1. A EP (passage 3) LP (passage 16) 200 µm 200 µm 1.5 3 B Mass spectrometry proteomics (n = 4) C mRNA (n = 4) D 100k EP 1.0 2 p < 0.0001 p < 0.0001 LP p < 0.0001 p < 0.0001 ) 0.5 1 2 p < 0.0001 p < 0.0001 10k 0.0 0 -0.5 -1 Cell area (µm Cell area fold change vs. EP fold change vs. -

Silencer of Death Domain (BAG4) (NM 004874) Human Recombinant Protein Product Data
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for TP306235 Silencer of Death Domain (BAG4) (NM_004874) Human Recombinant Protein Product data: Product Type: Recombinant Proteins Description: Recombinant protein of human BCL2-associated athanogene 4 (BAG4) Species: Human Expression Host: HEK293T Tag: C-Myc/DDK Predicted MW: 49.4 kDa Concentration: >50 ug/mL as determined by microplate BCA method Purity: > 80% as determined by SDS-PAGE and Coomassie blue staining Buffer: 25 mM Tris.HCl, pH 7.3, 100 mM glycine, 10% glycerol Preparation: Recombinant protein was captured through anti-DDK affinity column followed by conventional chromatography steps. Storage: Store at -80°C. Stability: Stable for 12 months from the date of receipt of the product under proper storage and handling conditions. Avoid repeated freeze-thaw cycles. RefSeq: NP_004865 Locus ID: 9530 UniProt ID: O95429 RefSeq Size: 4478 Cytogenetics: 8p11.23 RefSeq ORF: 1371 Synonyms: BAG-4; SODD This product is to be used for laboratory only. Not for diagnostic or therapeutic use. View online » ©2021 OriGene Technologies, Inc., 9620 Medical Center Drive, Ste 200, Rockville, MD 20850, US 1 / 2 Silencer of Death Domain (BAG4) (NM_004874) Human Recombinant Protein – TP306235 Summary: The protein encoded by this gene is a member of the BAG1-related protein family. BAG1 is an anti-apoptotic protein that functions through interactions with a variety of cell apoptosis and growth related proteins including BCL-2, Raf-protein kinase, steroid hormone receptors, growth factor receptors and members of the heat shock protein 70 kDa family. -

Bcl‑2 Associated Athanogene 4 Promotes Proliferation, Migration and Invasion of Gastric Cancer Cells
MOLECULAR MEDICINE REPORTS 16: 3753-3760, 2017 Bcl‑2 associated athanogene 4 promotes proliferation, migration and invasion of gastric cancer cells LIZHI YI1*, ZHENBING LV2,3*, JIANMEI WANG4 and XIANFEI ZHONG1 1Department of Gastroenterology, Leshan People's Hospital, Leshan, Sichuan 614000; 2Department of General Surgery Two, Nanchong Central Hospital; 3The Second Clinical School of North Sichuan Medical College, Nanchong, Sichuan 637000; 4Department of Pathology, The Affiliated Hospital of Southwest Medical University, Luzhou, Sichuan 646000, P.R. China Received May 22, 2016; Accepted April 4, 2017 DOI: 10.3892/mmr.2017.7073 Abstract. Currently, with the increase of morbidity and the rate of GC mortality has been decreased (4). It is necessary mortality rate, gastric cancer (GC) is attracting increasing to identify the biomarkers that can distinguish between GC attention in China. Bcl-2-associated athanogene 4 (BAG4) patients with poor or good prognosis. has been identified as a tumor promoter in several tumors, Bcl-2 associated athanogene 4 [BAG4, also known as but its role in GC remains unknown. The present study Silencer of Death Domains (SODD)] is a member of the aimed to detect the expression of BAG4 and determine its BAG1‑related protein family (5). The BAG proteins are located function in the progression of GC. The results from reverse in both cytoplasmic and nuclear in cells (6), although the func- transcription‑quantitative polymerase chain reaction and tional importance of this remains unclear. BAG proteins have a western blotting revealed that BAG4 was markedly upregu- conserved BAG domain (BD) that binds to the ATPase domain lated in highly metastatic cell lines (SGC7901 and MGC803), of Hsp70/Hsc70, and regulates the activity of these molecular compared with the lower‑metastatic cell lines (AGS and chaperones (7-9). -

Reassessing Apoptosis in Plants
PERSPECTIVE DOI: 10.1038/s41477-017-0020-x Reassessing apoptosis in plants Martin Dickman1,2*, Brett Williams3*, Yurong Li1,2, Paul Figueiredo1,4,5 and Thomas Wolpert6,7 Cell death can be driven by a genetically programmed signalling pathway known as programmed cell death (PCD). In plants, PCD occurs during development as well as in response to environmental and biotic stimuli. Our understanding of PCD regulation in plants has advanced significantly over the past two decades; however, the molecular machinery responsible for driving the system remains elusive. Thus, whether conserved PCD regulatory mechanisms include plant apoptosis remains enigmatic. Animal apoptotic regulators, including Bcl-2 family members, have not been identified in plants but expression of such regulators can trigger or suppress plant PCD. Moreover, plants exhibit nearly all of the biochemical and morphological features of apoptosis. One difference between plant and animal PCD is the absence of phagocytosis in plants. Evidence is emerging that the vacuole may be key to removal of unwanted plant cells, and may carry out functions that are analogous to animal phagocytosis. Here, we provide context for the argument that apoptotic-like cell death occurs in plants. ulticellular organisms possess intrinsic and extrinsic neurodegenerative diseases, such as Alzheimer’s and Parkinson’s programs for cell suicide. Programmed cell death (PCD) (too much PCD)16,17. Mis genetically controlled either by so-called death receptors Apoptosis is the most intensively studied form of PCD and in or intrinsic signals that direct the cell to eliminate itself during animals occurs by two major pathways, extrinsic and intrinsic, development and in response to pathogens or environmental distinguished by the origin of the death signal. -

T Cell Receptor Signaling Pathway and Cytokine-Cytokine Receptor Interaction Affect the Rehabilitation Process After Respiratory Syncytial Virus Infection
T cell receptor signaling pathway and cytokine-cytokine receptor interaction affect the rehabilitation process after respiratory syncytial virus infection Zuanhao Qian*, Zhenglei Zhang* and Yingying Wang Department of Pediatrics, Taikang Xianlin Drum Tower Hospital, Nanjing, China * These authors contributed equally to this work. ABSTRACT Background. Respiratory syncytial virus (RSV) is the main cause of respiratory tract infection, which seriously threatens the health and life of children. This study is conducted to reveal the rehabilitation mechanisms of RSV infection. Methods. E-MTAB-5195 dataset was downloaded from EBI ArrayExpress database, including 39 acute phase samples in the acute phase of infection and 21 samples in the recovery period. Using the limma package, differentially expressed RNAs (DE- RNAs) were analyzed. The significant modules were identified using WGCNA package, and the mRNAs in them were conducted with enrichment analysis using DAVID tool. Afterwards, co-expression network for the RNAs involved in the significant modules was built by Cytoscape software. Additionally, RSV-correlated pathways were searched from Comparative Toxicogenomics Database, and then the pathway network was constructed. Results. There were 2,489 DE-RNAs between the two groups, including 2,386 DE- mRNAs and 103 DE-lncRNAs. The RNAs in the black, salmon, blue, tan and turquoise modules correlated with stage were taken as RNA set1. Meanwhile, the RNAs in brown, blue, magenta and pink modules related to disease severity were defined as RNA set2. In the pathway networks, CD40LG and RASGRP1 co-expressed Submitted 14 January 2019 with LINC00891/LINC00526/LINC01215 were involved in the T cell receptor sig- Accepted 6 May 2019 Published 12 June 2019 naling pathway, and IL1B, IL1R2, IL18, and IL18R1 co-expressed with BAIAP2- AS1/CRNDE/LINC01503/SMIM25 were implicated in cytokine-cytokine receptor Corresponding author Zuanhao Qian, interaction. -

RESEARCH ARTICLE Evaluation of Apoptotic Gene Expression In
DOI:10.31557/APJCP.2021.22.5.1413 Apoptotic Activity of Nisin RESEARCH ARTICLE Editorial Process: Submission:00/00/0000 Acceptance:00/00/0000 Evaluation of Apoptotic Gene Expression in Hepatoma Cell Line (HepG2) Following Nisin Treatment Nahid Zainodini1, Mohammad Reza Hajizadeh2,3, Mohammad Reza Mirzaei2,3* Abstract Objective: The present study aims to examine the effects of nisin on the survival and apoptosis of the hepatoma cell line HepG2 and to investigate possible apoptosis pathways activated by nisin. Materials and Methods: For this purpose, viability and apoptosis of the cells were accomplished by the nisin treatment using the MTT assay and Annexin- V-fluorescein/propidium iodide (PI) double staining, respectively. Additionally, the human apoptosis PCR array was performed to determine pathways or genes activated by nisin during possible apoptosis. Results: The results of the present study showed that nisin was able to decrease cell viability (IC50 ~ 40 µg/ml) in a dose-dependent manner and could induce apoptosis in HepG2 cells. PCR data indicated a considerable increase in the expression of genes, such as caspase and BCL2 families, involved in the induction of apoptosis. Conclusions: The data from this study showed that overexpression of genes involved in the intrinsic pathway of apoptosis, especially caspase-9 and BID, increased apoptosis in HepG2 cells treated by nisin, compared to the control group. Keywords: Liver cancer- antimicrobial peptide- programmed cell death- cancer treatmen- nisin Asian Pac J Cancer Prev, 22 (5), 1413-1419 Introduction development of complex multicellular organisms (Zasloff, 2002). Nisin is an antimicrobial peptide composed of Hepatocellular carcinoma is the third-most prevalent particular gram-positive bacteria, such as the species cause of cancer-related deaths leading to approximately Streptococcus and Lactococcus (Lubelski et al., 2009). -

Supplemental Solier
Supplementary Figure 1. Importance of Exon numbers for transcript downregulation by CPT Numbers of down-regulated genes for four groups of comparable size genes, differing only by the number of exons. Supplementary Figure 2. CPT up-regulates the p53 signaling pathway genes A, List of the GO categories for the up-regulated genes in CPT-treated HCT116 cells (p<0.05). In bold: GO category also present for the genes that are up-regulated in CPT- treated MCF7 cells. B, List of the up-regulated genes in both CPT-treated HCT116 cells and CPT-treated MCF7 cells (CPT 4 h). C, RT-PCR showing the effect of CPT on JUN and H2AFJ transcripts. Control cells were exposed to DMSO. β2 microglobulin (β2) mRNA was used as control. Supplementary Figure 3. Down-regulation of RNA degradation-related genes after CPT treatment A, “RNA degradation” pathway from KEGG. The genes with “red stars” were down- regulated genes after CPT treatment. B, Affy Exon array data for the “CNOT” genes. The log2 difference for the “CNOT” genes expression depending on CPT treatment was normalized to the untreated controls. C, RT-PCR showing the effect of CPT on “CNOT” genes down-regulation. HCT116 cells were treated with CPT (10 µM, 20 h) and CNOT6L, CNOT2, CNOT4 and CNOT6 mRNA were analysed by RT-PCR. Control cells were exposed to DMSO. β2 microglobulin (β2) mRNA was used as control. D, CNOT6L down-regulation after CPT treatment. CNOT6L transcript was analysed by Q- PCR. Supplementary Figure 4. Down-regulation of ubiquitin-related genes after CPT treatment A, “Ubiquitin-mediated proteolysis” pathway from KEGG. -

High HSPA8 Expression Predicts Adverse Outcomes of Acute Myeloid Leukemia Jun Li1 and Zheng Ge1,2*
Li and Ge BMC Cancer (2021) 21:475 https://doi.org/10.1186/s12885-021-08193-w RESEARCH Open Access High HSPA8 expression predicts adverse outcomes of acute myeloid leukemia Jun Li1 and Zheng Ge1,2* Abstract Background: Acute myeloid leukemia (AML) remains one of the most common hematological malignancies, posing a serious challenge to human health. HSPA8 is a chaperone protein that facilitates proper protein folding. It contributes to various activities of cell function and also is associated with various types of cancers. To date, the role of HSPA8 in AML is still undetermined. Methods: In this study, public datasets available from the TCGA (Cancer Genome Atlas) and GEO (Gene Expression Omnibus) were mined to discover the association between the expression of HSPA8 and clinical phenotypes of CN- AML. A series of bioinformatics analysis methods, including functional annotation and miRNA-mRNA regulation network analysis, were employed to investigate the role of HSPA8 in CN-AML. Results: HSPA8 was highly expressed in the AML patients compared to the healthy controls. The high HSPA8 expression had lower overall survival (OS) rate than those with low HSPA8 expression. High expression of HSPA8 was also an independent prognostic factor for overall survival (OS) of CN-AML patients by multivariate analysis. The differential expressed genes (DEGs) associated with HSPA8 high expression were identified, and they were enriched PI3k-Akt signaling, cAMP signaling, calcium signaling pathway. HSPA8 high expression was also positively associated with micro-RNAs (hsa-mir-1269a, hsa-mir-508-3p, hsa-mir-203a), the micro-RNAs targeted genes (VSTM4, RHOB, HOBX7) and key known oncogenes (KLF5, RAN, and IDH1), and negatively associated with tumor suppressors (KLF12, PRKG1, TRPS1, NOTCH1, RORA). -

Silencer of Death Domain (BAG4) (NM 004874) Human Untagged Clone Product Data
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for SC125513 Silencer of Death Domain (BAG4) (NM_004874) Human Untagged Clone Product data: Product Type: Expression Plasmids Product Name: Silencer of Death Domain (BAG4) (NM_004874) Human Untagged Clone Tag: Tag Free Symbol: BAG4 Synonyms: BAG-4; SODD Vector: pCMV6-XL4 E. coli Selection: Ampicillin (100 ug/mL) Cell Selection: None Fully Sequenced ORF: >OriGene ORF within SC125513 sequence for NM_004874 edited (data generated by NextGen Sequencing) ATGTCGGCCCTGAGGCGCTCGGGCTACGGCCCCAGTGACGGTCCGTCCTACGGCCGCTAC TACGGGCCTGGGGGTGGAGATGTGCCGGTACACCCACCTCCACCCTTATATCCTCTTCGC CCTGAACCTCCCCAGCCTCCCATTTCCTGGCGGGTGCGCGGGGGCGGCCCGGCGGAGACC ACCTGGCTGGGAGAAGGCGGAGGAGGCGATGGCTACTATCCCTCGGGAGGCGCCTGGCCA GAGCCTGGTCGAGCCGGAGGAAGCCACCAGGAGCAGCCACCATATCCTAGCTACAATTCT AACTATTGGAATTCTACTGCGAGATCTAGGGCTCCTTACCCAAGTACATATCCTGTAAGA CCAGAATTGCAAGGCCAGAGTTTGAATTCTTATACAAATGGAGCGTATGGTCCAACATAC CCCCCAGGCCCTGGGGCAAATACTGCCTCATACTCAGGGGCTTATTATGCACCTGGTTAT ACTCAGACCAGTTACTCCACAGAAGTTCCAAGTACTTACCGTTCATCTGGCAACAGCCCA ACTCCAGTCTCTCGTTGGATCTATCCCCAGCAGGACTGTCAGACTGAAGCACCCCCTCTT AGGGGGCAGGTTCCAGGATATCCGCCTTCACAGAACCCTGGAATGACCCTGCCCCATTAT CCTTATGGAGATGGTAATCGTAGTGTTCCACAATCAGGACCGACTGTACGACCACAAGAA GATGCGTGGGCTTCTCCTGGTGCTTATGGAATGGGTGGCCGTTATCCCTGGCCTTCATCA GCGCCCTCAGCACCACCCGGCAATCTCTACATGACTGAAAGTACTTCACCATGGCCTAGC AGTGGCTCTCCCCAGTCACCCCCTTCACCCCCAGTCCAGCAGCCCAAGGATTCTTCATAC