Molecular Chaperones As Regulators of Cell Death
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

HAC-1, a Drosophila Homolog of APAF-1 and CED-4, Functions in Developmental and Radiation-Induced Apoptosis
Molecular Cell, Vol. 4, 745±755, November, 1999, Copyright 1999 by Cell Press HAC-1, a Drosophila Homolog of APAF-1 and CED-4, Functions in Developmental and Radiation-Induced Apoptosis Lei Zhou, Zhiwei Song,² Jan Tittel,² generation of a large (p20) and small (p10) subunit. Cas- and Hermann Steller* pases can be activated through cleavage by active ªiniti- Howard Hughes Medical Institute atorº caspases in a caspase cascade (Li et al., 1997), Department of Biology and by autoproteolysis following aggregation of two or Massachusetts Institute of Technology more zymogen molecules (MacCorkle et al., 1998; Muzio Cambridge, Massachusetts 02139 et al., 1998; Yang et al., 1998). Upon activation of ªexecu- tionerº caspases, a wide variety of specific intracellular proteins are cleaved in different cellular compartments, and it is thought that their breakdown ultimately leads to Summary the characteristic morphological changes of apoptosis (Nicholson and Thornberry, 1997). The activation of cas- We have identified a Drosophila homolog of Apaf-1 pases appears to be tightly controlled by both positive and ced-4, termed hac-1. Like mammalian APAF-1, HAC-1 can activate caspases in a dATP-dependent and negative regulators. On the one hand, members of manner in vitro. During embryonic development, hac-1 the IAP (inhibitor of apoptosis protein) family can inhibit is prominently expressed in regions where cells un- caspases and apoptosis in a variety of insect and verte- dergo natural death. Significantly, hac-1 transcription brate systems (Uren et al., 1998; Deveraux and Reed, is also rapidly induced upon ionizing irradiation, similar 1999). On the other hand, caspase activation is stimu- to the proapoptotic gene reaper. -

Deletion of Endoplasmic Reticulum Stress-Responsive Co-Chaperone P58ipk Protects Mice from Diet-Induced Steatohepatitis
Zurich Open Repository and Archive University of Zurich Main Library Strickhofstrasse 39 CH-8057 Zurich www.zora.uzh.ch Year: 2018 Deletion of endoplasmic reticulum stress-responsive co-chaperone p58IPK protects mice from diet-induced steatohepatitis Bandla, Harikrishna ; Dasgupta, Debanjali ; Mauer, Amy S ; Nozickova, Barbora ; Kumar, Swarup ; Hirsova, Petra ; Graham, Rondell P ; Malhi, Harmeet DOI: https://doi.org/10.1111/hepr.13052 Posted at the Zurich Open Repository and Archive, University of Zurich ZORA URL: https://doi.org/10.5167/uzh-144890 Journal Article Accepted Version Originally published at: Bandla, Harikrishna; Dasgupta, Debanjali; Mauer, Amy S; Nozickova, Barbora; Kumar, Swarup; Hirsova, Petra; Graham, Rondell P; Malhi, Harmeet (2018). Deletion of endoplasmic reticulum stress-responsive co-chaperone p58IPK protects mice from diet-induced steatohepatitis. Hepatology Research, 48(6):479- 494. DOI: https://doi.org/10.1111/hepr.13052 Deletion of endoplasmic reticulum stress-responsive co-chaperone p58IPK protects mice from diet-induced steatohepatitis Harikrishna Bandla1, Debanjali Dasgupta1, Amy S. Mauer1, Barbora Nozickova2, Swarup Kumar3, Petra Hirsova1, Rondell P. Graham4, Harmeet Malhi1* 1. Division of Gastroenterology and Hepatology, Mayo Clinic, Rochester, MN 2. Universitatsspital Zurich, 8096, Ramistrasse 100, Zurich, Switzerland 3. Department of Medicine, Saint Vincent Hospital, 123 Summer St, Worcester, MA 4. Department of Laboratory Medicine and Pathology, Mayo Clinic, Rochester, MN Corresponding author: Harmeet Malhi, M.B.B.S. Associate Professor of Medicine and Physiology Mayo Clinic College of Medicine 200 First Street SW Rochester, MN 55905 Tel: 507 284 0686 Fax: 507 284 0762 Email: [email protected] Funding: This work was supported by DK 97178, DK107402 and DK111378 (H.M.), the Robert and Elizabeth Strickland Career Development Award from the Division of Endocrinology (H.M.), the Gilead Sciences Research Scholars Program in Liver Disease (H.M.) and the Palumbo Foundation (H.M.), the Edward C. -
![Computational Genome-Wide Identification of Heat Shock Protein Genes in the Bovine Genome [Version 1; Peer Review: 2 Approved, 1 Approved with Reservations]](https://docslib.b-cdn.net/cover/8283/computational-genome-wide-identification-of-heat-shock-protein-genes-in-the-bovine-genome-version-1-peer-review-2-approved-1-approved-with-reservations-88283.webp)
Computational Genome-Wide Identification of Heat Shock Protein Genes in the Bovine Genome [Version 1; Peer Review: 2 Approved, 1 Approved with Reservations]
F1000Research 2018, 7:1504 Last updated: 08 AUG 2021 RESEARCH ARTICLE Computational genome-wide identification of heat shock protein genes in the bovine genome [version 1; peer review: 2 approved, 1 approved with reservations] Oyeyemi O. Ajayi1,2, Sunday O. Peters3, Marcos De Donato2,4, Sunday O. Sowande5, Fidalis D.N. Mujibi6, Olanrewaju B. Morenikeji2,7, Bolaji N. Thomas 8, Matthew A. Adeleke 9, Ikhide G. Imumorin2,10,11 1Department of Animal Breeding and Genetics, Federal University of Agriculture, Abeokuta, Nigeria 2International Programs, College of Agriculture and Life Sciences, Cornell University, Ithaca, NY, 14853, USA 3Department of Animal Science, Berry College, Mount Berry, GA, 30149, USA 4Departamento Regional de Bioingenierias, Tecnologico de Monterrey, Escuela de Ingenieria y Ciencias, Queretaro, Mexico 5Department of Animal Production and Health, Federal University of Agriculture, Abeokuta, Nigeria 6Usomi Limited, Nairobi, Kenya 7Department of Animal Production and Health, Federal University of Technology, Akure, Nigeria 8Department of Biomedical Sciences, Rochester Institute of Technology, Rochester, NY, 14623, USA 9School of Life Sciences, University of KwaZulu-Natal, Durban, 4000, South Africa 10School of Biological Sciences, Georgia Institute of Technology, Atlanta, GA, 30032, USA 11African Institute of Bioscience Research and Training, Ibadan, Nigeria v1 First published: 20 Sep 2018, 7:1504 Open Peer Review https://doi.org/10.12688/f1000research.16058.1 Latest published: 20 Sep 2018, 7:1504 https://doi.org/10.12688/f1000research.16058.1 Reviewer Status Invited Reviewers Abstract Background: Heat shock proteins (HSPs) are molecular chaperones 1 2 3 known to bind and sequester client proteins under stress. Methods: To identify and better understand some of these proteins, version 1 we carried out a computational genome-wide survey of the bovine 20 Sep 2018 report report report genome. -

The HECT Domain Ubiquitin Ligase HUWE1 Targets Unassembled Soluble Proteins for Degradation
OPEN Citation: Cell Discovery (2016) 2, 16040; doi:10.1038/celldisc.2016.40 ARTICLE www.nature.com/celldisc The HECT domain ubiquitin ligase HUWE1 targets unassembled soluble proteins for degradation Yue Xu1, D Eric Anderson2, Yihong Ye1 1Laboratory of Molecular Biology, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, MD, USA; 2Advanced Mass Spectrometry Core Facility, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, MD, USA In eukaryotes, many proteins function in multi-subunit complexes that require proper assembly. To maintain complex stoichiometry, cells use the endoplasmic reticulum-associated degradation system to degrade unassembled membrane subunits, but how unassembled soluble proteins are eliminated is undefined. Here we show that degradation of unassembled soluble proteins (referred to as unassembled soluble protein degradation, USPD) requires the ubiquitin selective chaperone p97, its co-factor nuclear protein localization protein 4 (Npl4), and the proteasome. At the ubiquitin ligase level, the previously identified protein quality control ligase UBR1 (ubiquitin protein ligase E3 component n-recognin 1) and the related enzymes only process a subset of unassembled soluble proteins. We identify the homologous to the E6-AP carboxyl terminus (homologous to the E6-AP carboxyl terminus) domain-containing protein HUWE1 as a ubiquitin ligase for substrates bearing unshielded, hydrophobic segments. We used a stable isotope labeling with amino acids-based proteomic approach to identify endogenous HUWE1 substrates. Interestingly, many HUWE1 substrates form multi-protein com- plexes that function in the nucleus although HUWE1 itself is cytoplasmically localized. Inhibition of nuclear entry enhances HUWE1-mediated ubiquitination and degradation, suggesting that USPD occurs primarily in the cytoplasm. -

Identification of the Binding Partners for Hspb2 and Cryab Reveals
Brigham Young University BYU ScholarsArchive Theses and Dissertations 2013-12-12 Identification of the Binding arP tners for HspB2 and CryAB Reveals Myofibril and Mitochondrial Protein Interactions and Non- Redundant Roles for Small Heat Shock Proteins Kelsey Murphey Langston Brigham Young University - Provo Follow this and additional works at: https://scholarsarchive.byu.edu/etd Part of the Microbiology Commons BYU ScholarsArchive Citation Langston, Kelsey Murphey, "Identification of the Binding Partners for HspB2 and CryAB Reveals Myofibril and Mitochondrial Protein Interactions and Non-Redundant Roles for Small Heat Shock Proteins" (2013). Theses and Dissertations. 3822. https://scholarsarchive.byu.edu/etd/3822 This Thesis is brought to you for free and open access by BYU ScholarsArchive. It has been accepted for inclusion in Theses and Dissertations by an authorized administrator of BYU ScholarsArchive. For more information, please contact [email protected], [email protected]. Identification of the Binding Partners for HspB2 and CryAB Reveals Myofibril and Mitochondrial Protein Interactions and Non-Redundant Roles for Small Heat Shock Proteins Kelsey Langston A thesis submitted to the faculty of Brigham Young University in partial fulfillment of the requirements for the degree of Master of Science Julianne H. Grose, Chair William R. McCleary Brian Poole Department of Microbiology and Molecular Biology Brigham Young University December 2013 Copyright © 2013 Kelsey Langston All Rights Reserved ABSTRACT Identification of the Binding Partners for HspB2 and CryAB Reveals Myofibril and Mitochondrial Protein Interactors and Non-Redundant Roles for Small Heat Shock Proteins Kelsey Langston Department of Microbiology and Molecular Biology, BYU Master of Science Small Heat Shock Proteins (sHSP) are molecular chaperones that play protective roles in cell survival and have been shown to possess chaperone activity. -
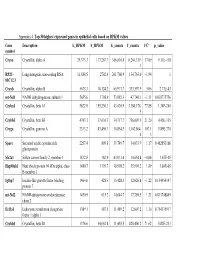
Appendix 4. Top 50 Highest Expressed Genes in Epithelial Cells Based on RPKM Values
Appendix 4. Top 50 highest expressed genes in epithelial cells based on RPKM values Gene Description E_RPKM F_RPKM E_counts F_counts FC* p_value symbol Cryaa Crystallin, alpha A 29,373.3 177,267.7 366,616.4 6,264,319. 17.09 9.11E-118 1 RP23– Long intergenic non-coding RNA 11,888.5 2702.4 261,760.9 134,763.0 −1.94 1 81C12.3 Cryab Crystallin, alpha B 5673.3 10,124.2 65,971.7 333,597.9 5.06 2.71E-43 mt-Nd1 NADH dehydrogenase, subunit 1 5655.6 1798.9 53,082.3 47,748.1 −1.11 0.838775756 Cryba1 Crystallin, beta A1 5622.0 155,230.3 43,420.9 3,380,176. 77.85 1.34E-240 5 Crybb3 Crystallin, beta B3 4743.1 37,636.3 34,717.7 736,007.9 21.20 4.45E-135 Cryga Crystallin, gamma A 2333.2 83,496.3 10,854.5 1,162,864. 107.1 5.89E-270 6 3 Sparc Secreted acidic cysteine rich 2257.4 809.8 39,749.7 34,033.9 −1.17 0.462853166 glycoprotein Slc2a1 Solute carrier family 2, member 1 1832.8 162.9 43,031.4 10,654.8 −4.04 1.67E-05 Hsp90ab1 Heat shock protein 90 kDa alpha, class 1480.7 1139.7 18,998.2 35,901.2 1.89 3.84E-05 B member 1 Igfbp7 Insulin-like growth factor binding 1464.6 428.3 15,428.3 12,626.8 −1.22 0.154954147 protein 7 mt-Nd2 NADH-ubiquinone oxidoreductase 1450.9 615.2 14,644.7 17,789.5 1.21 0.833748849 chain 2 Eef1a1 Eukaryotic translation elongation 1389.1 587.5 11,489.2 12,607.2 1.10 0.754135917 factor 1 alpha 1 Crybb1 Crystallin, beta B1 1376.6 34,662.8 11,455.5 820,406.2 71.62 5.82E-233 Htra3 HtrA serine peptidase 3 1338.6 162.0 23,197.6 6433.9 −3.61 3.93E-05 Gnb2l1 Guanine nucleotide-binding protein 1293.3 670.1 14,495.1 21,652.1 1.49 0.001685952 -

Apoptosis Induced by Proteasome Inhibition in Cancer Cells: Predominant Role of the P53/PUMA Pathway
Oncogene (2007) 26, 1681–1692 & 2007 Nature Publishing Group All rights reserved 0950-9232/07 $30.00 www.nature.com/onc ORIGINAL ARTICLE Apoptosis induced by proteasome inhibition in cancer cells: predominant role of the p53/PUMA pathway CG Concannon1, BF Koehler1,2, Claus Reimertz2, BM Murphy1, C Bonner1, N Thurow2, MW Ward1, AVillunger 3, AStrasser 4,DKo¨ gel2,5 and JHM Prehn1,5 1Department of Physiology and Medical Physics, Royal College of Surgeons in Ireland, Dublin, Ireland; 2Experimental Neurosurgery, Centre for Neurology and Neurosurgery, Johann Wolfgang Goethe University Clinics, Theodor-Stern-Kai 7, Frankfurt/Main, Germany; 3Division of Experimental Pathophysiology and Immunology, Biocenter, Innsbruck Medical University, Innsbruck, Austria and 4The Walter and Eliza Hall Institute of Medical Research, Melbourne, Australia The proteasome has emerged as a novel target for Introduction antineoplastic treatment of hematological malignancies and solid tumors, including those of the central nervous The correct functioning of the ubiquitin-proteasome system. To identify cell death pathways activated in pathway is essential for the degradation of the majority response to inhibition of the proteasome system in cancer of intracellular proteins. Several key regulatory proteins cells, we treated human SH-SY5Y neuroblastoma cells involved in cell proliferation and differentiation are with the selective proteasome inhibitor (PI) epoxomicin regulated by proteasome-mediated proteolysis resulting (Epoxo). Prolonged exposure to Epoxo was associated in the activation or inhibition of specific cell signaling with increased levels of poly-ubiquitinylated proteins and pathways (Adams, 2004a). The proteasome is also p53, release of cytochrome c from the mitochondria, and central to the regulation of cell death and apoptosis. -

The Role of RNA Editing in Cancer Development and Metabolic Disorders
Washington University School of Medicine Digital Commons@Becker Open Access Publications 2018 The oler of RNA editing in cancer development and metabolic disorders Che-Pei Kung Washington University School of Medicine in St. Louis Leonard B. Maggi Jr. Washington University School of Medicine in St. Louis Jason D. Weber Washington University School of Medicine in St. Louis Follow this and additional works at: https://digitalcommons.wustl.edu/open_access_pubs Recommended Citation Kung, Che-Pei; Maggi, Leonard B. Jr.; and Weber, Jason D., ,"The or le of RNA editing in cancer development and metabolic disorders." Frontiers in endocrinology.9,. 762. (2018). https://digitalcommons.wustl.edu/open_access_pubs/7400 This Open Access Publication is brought to you for free and open access by Digital Commons@Becker. It has been accepted for inclusion in Open Access Publications by an authorized administrator of Digital Commons@Becker. For more information, please contact [email protected]. REVIEW published: 18 December 2018 doi: 10.3389/fendo.2018.00762 The Role of RNA Editing in Cancer Development and Metabolic Disorders Che-Pei Kung 1,2*, Leonard B. Maggi Jr. 1,2 and Jason D. Weber 1,2,3* 1 ICCE Institute, Washington University School of Medicine, Saint Louis, MO, United States, 2 Division of Molecular Oncology, Department of Medicine, Washington University School of Medicine, Saint Louis, MO, United States, 3 Siteman Cancer Center, Department of Cell Biology and Physiology, Washington University School of Medicine, Saint Louis, MO, United States Numerous human diseases arise from alterations of genetic information, most notably DNA mutations. Thought to be merely the intermediate between DNA and protein, changes in RNA sequence were an afterthought until the discovery of RNA editing 30 years ago. -

Heat Shock Protein 27 Inhibits HMGB1 Translocation by Regulating CBP
Molecular Immunology 108 (2019) 45–55 Contents lists available at ScienceDirect Molecular Immunology journal homepage: www.elsevier.com/locate/molimm Heat shock protein 27 inhibits HMGB1 translocation by regulating CBP acetyltransferase activity and ubiquitination T ⁎⁎ Xiaowen Bia, Miao Xua, Jinfei Lia, Ting Huanga, Baolin Jianga, Lei Shena, Lan Luob, , ⁎⁎⁎ ⁎ Shixiang Liuc, , Zhimin Yina, a Jiangsu Province Key Laboratory for Molecular and Medical Biotechnology, College of Life Science, Nanjing Normal University, Nanjing, Jiangsu, PR China b State Key Laboratory of Pharmaceutical Biotechnology, School of Life Sciences, Nanjing University, Nanjing, Jiangsu, PR China c Jurong People’s Hospital, Zhenjiang, Jiangsu, PR China ARTICLE INFO ABSTRACT Keywords: Heat-shock protein 27 (Hsp27) is a member of the small heat shock protein family that has been reported to Hsp27 protect cells against pro-inflammatory stresses. High mobility group box 1 (HMGB1) is a proinflammatory cy- CBP tokine associated with death from sepsis and other inflammatory diseases. After being acetylated by CREB- HMGB1 binding protein (CBP), the transcriptional adaptor and acetyltransferase, HMGB1 translocates from the nucleus Phosphorylation to the cytoplasm. In the present study, we investigated the effects of Hsp27 on HMGB1 translocation from the Acetylation nucleus to the cytoplasm in THP-1 cells. We found that Hsp27 phosphorylation decreased LPS-induced HMGB1 acetylation and translocation from the nucleus to the cytoplasm, as well as its release from THP-1 cells. The study further showed that cytosolic non-phosphorylated Hsp27 enhanced CBP ubiquitination and degradation in LPS-unstimulated cells, which suggested that Hsp27 maintained suitable CBP levels under normal physiological conditions. After LPS stimulation, Hsp27 was phosphorylated at serine residues 15/78 and translocated from the cytoplasm into the nucleus. -

Figure S1. DMD Module Network. the Network Is Formed by 260 Genes from Disgenet and 1101 Interactions from STRING. Red Nodes Are the Five Seed Candidate Genes
Figure S1. DMD module network. The network is formed by 260 genes from DisGeNET and 1101 interactions from STRING. Red nodes are the five seed candidate genes. Figure S2. DMD module network is more connected than a random module of the same size. It is shown the distribution of the largest connected component of 10.000 random modules of the same size of the DMD module network. The green line (x=260) represents the DMD largest connected component, obtaining a z-score=8.9. Figure S3. Shared genes between BMD and DMD signature. A) A meta-analysis of three microarray datasets (GSE3307, GSE13608 and GSE109178) was performed for the identification of differentially expressed genes (DEGs) in BMD muscle biopsies as compared to normal muscle biopsies. Briefly, the GSE13608 dataset included 6 samples of skeletal muscle biopsy from healthy people and 5 samples from BMD patients. Biopsies were taken from either biceps brachii, triceps brachii or deltoid. The GSE3307 dataset included 17 samples of skeletal muscle biopsy from healthy people and 10 samples from BMD patients. The GSE109178 dataset included 14 samples of controls and 11 samples from BMD patients. For both GSE3307 and GSE10917 datasets, biopsies were taken at the time of diagnosis and from the vastus lateralis. For the meta-analysis of GSE13608, GSE3307 and GSE109178, a random effects model of effect size measure was used to integrate gene expression patterns from the two datasets. Genes with an adjusted p value (FDR) < 0.05 and an │effect size│>2 were identified as DEGs and selected for further analysis. A significant number of DEGs (p<0.001) were in common with the DMD signature genes (blue nodes), as determined by a hypergeometric test assessing the significance of the overlap between the BMD DEGs and the number of DMD signature genes B) MCODE analysis of the overlapping genes between BMD DEGs and DMD signature genes. -

HDAC Inhibition Activates the Apoptosome Via Apaf1 Upregulation
Buurman et al. Eur J Med Res (2016) 21:26 DOI 10.1186/s40001-016-0217-x European Journal of Medical Research RESEARCH Open Access HDAC inhibition activates the apoptosome via Apaf1 upregulation in hepatocellular carcinoma Reena Buurman, Maria Sandbothe, Brigitte Schlegelberger and Britta Skawran* Abstract Background: Histone deacetylation, a common hallmark in malignant tumors, strongly alters the transcription of genes involved in the control of proliferation, cell survival, differentiation and genetic stability. We have previously shown that HDAC1, HDAC2, and HDAC3 (HDAC1–3) genes encoding histone deacetylases 1–3 are upregulated in primary human hepatocellular carcinoma (HCC). The aim of this study was to characterize the functional effects of HDAC1–3 downregulation and to identify functionally important target genes of histone deacetylation in HCC. Methods: Therefore, HCC cell lines were treated with the histone deacetylase inhibitor (HDACi) trichostatin A and by siRNA-knockdown of HDAC1–3. Differentially expressed mRNAs were identified after siRNA-knockdown of HDAC1–3 using mRNA expression profiling. Findings were validated after siRNA-mediated silencing of HDAC1–3 using qRTPCR and Western blotting assays. Results: mRNA profiling identified apoptotic protease-activating factor 1 (Apaf1) to be significantly upregulated after HDAC inhibition (HLE siRNA#1/siRNA#2 p < 0.05, HLF siRNA#1/siRNA#2 p < 0.05). As a component of the apoptosome, a caspase-activating complex, Apaf1 plays a central role in the mitochondrial caspase activation pathway of apopto- sis. Using annexin V, a significant increase in apoptosis could also be shown in HLE (siRNA #1 p 0.0034) and HLF after siRNA against HDAC1–3 (Fig. -

Apoptotic Threshold Is Lowered by P53 Transactivation of Caspase-6
Apoptotic threshold is lowered by p53 transactivation of caspase-6 Timothy K. MacLachlan*† and Wafik S. El-Deiry*‡§¶ʈ** *Laboratory of Molecular Oncology and Cell Cycle Regulation, Howard Hughes Medical Institute, and Departments of ‡Medicine, §Genetics, ¶Pharmacology, and Cancer Center, University of Pennsylvania School of Medicine, Philadelphia, PA 19104 Communicated by Britton Chance, University of Pennsylvania School of Medicine, Philadelphia, PA, April 23, 2002 (received for review January 11, 2002) Little is known about how a cell’s apoptotic threshold is controlled Inhibition of the enzyme reduces the sensitivity conferred by after exposure to chemotherapy, although the p53 tumor suppres- overexpression of p53. These results identify a pathway by which sor has been implicated. We identified executioner caspase-6 as a p53 is able to accelerate the apoptosis cascade by loading the cell transcriptional target of p53. The mechanism involves DNA binding with cell death proteases so that when an apoptotic signal is by p53 to the third intron of the caspase-6 gene and transactiva- received, programmed cell death occurs rapidly. tion. A p53-dependent increase in procaspase-6 protein level al- lows for an increase in caspase-6 activity and caspase-6-specific Materials and Methods Lamin A cleavage in response to Adriamycin exposure. Specific Western Blotting and Antibodies. Immunoblotting was carried out by inhibition of caspase-6 blocks cell death in a manner that correlates using mouse anti-human p53 monoclonal (PAb1801; Oncogene), with caspase-6 mRNA induction by p53 and enhances long-term rabbit anti-human caspase-3 (Cell Signaling, Beverly, MA), mouse survival in response to a p53-mediated apoptotic signal.