Cranberry Proanthocyanidins Mediate Growth Arrest of Lung Cancer Cells Through Modulation of Gene Expression and Rapid Induction of Apoptosis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Seq2pathway Vignette
seq2pathway Vignette Bin Wang, Xinan Holly Yang, Arjun Kinstlick May 19, 2021 Contents 1 Abstract 1 2 Package Installation 2 3 runseq2pathway 2 4 Two main functions 3 4.1 seq2gene . .3 4.1.1 seq2gene flowchart . .3 4.1.2 runseq2gene inputs/parameters . .5 4.1.3 runseq2gene outputs . .8 4.2 gene2pathway . 10 4.2.1 gene2pathway flowchart . 11 4.2.2 gene2pathway test inputs/parameters . 11 4.2.3 gene2pathway test outputs . 12 5 Examples 13 5.1 ChIP-seq data analysis . 13 5.1.1 Map ChIP-seq enriched peaks to genes using runseq2gene .................... 13 5.1.2 Discover enriched GO terms using gene2pathway_test with gene scores . 15 5.1.3 Discover enriched GO terms using Fisher's Exact test without gene scores . 17 5.1.4 Add description for genes . 20 5.2 RNA-seq data analysis . 20 6 R environment session 23 1 Abstract Seq2pathway is a novel computational tool to analyze functional gene-sets (including signaling pathways) using variable next-generation sequencing data[1]. Integral to this tool are the \seq2gene" and \gene2pathway" components in series that infer a quantitative pathway-level profile for each sample. The seq2gene function assigns phenotype-associated significance of genomic regions to gene-level scores, where the significance could be p-values of SNPs or point mutations, protein-binding affinity, or transcriptional expression level. The seq2gene function has the feasibility to assign non-exon regions to a range of neighboring genes besides the nearest one, thus facilitating the study of functional non-coding elements[2]. Then the gene2pathway summarizes gene-level measurements to pathway-level scores, comparing the quantity of significance for gene members within a pathway with those outside a pathway. -

The Role of RNA Editing in Cancer Development and Metabolic Disorders
Washington University School of Medicine Digital Commons@Becker Open Access Publications 2018 The oler of RNA editing in cancer development and metabolic disorders Che-Pei Kung Washington University School of Medicine in St. Louis Leonard B. Maggi Jr. Washington University School of Medicine in St. Louis Jason D. Weber Washington University School of Medicine in St. Louis Follow this and additional works at: https://digitalcommons.wustl.edu/open_access_pubs Recommended Citation Kung, Che-Pei; Maggi, Leonard B. Jr.; and Weber, Jason D., ,"The or le of RNA editing in cancer development and metabolic disorders." Frontiers in endocrinology.9,. 762. (2018). https://digitalcommons.wustl.edu/open_access_pubs/7400 This Open Access Publication is brought to you for free and open access by Digital Commons@Becker. It has been accepted for inclusion in Open Access Publications by an authorized administrator of Digital Commons@Becker. For more information, please contact [email protected]. REVIEW published: 18 December 2018 doi: 10.3389/fendo.2018.00762 The Role of RNA Editing in Cancer Development and Metabolic Disorders Che-Pei Kung 1,2*, Leonard B. Maggi Jr. 1,2 and Jason D. Weber 1,2,3* 1 ICCE Institute, Washington University School of Medicine, Saint Louis, MO, United States, 2 Division of Molecular Oncology, Department of Medicine, Washington University School of Medicine, Saint Louis, MO, United States, 3 Siteman Cancer Center, Department of Cell Biology and Physiology, Washington University School of Medicine, Saint Louis, MO, United States Numerous human diseases arise from alterations of genetic information, most notably DNA mutations. Thought to be merely the intermediate between DNA and protein, changes in RNA sequence were an afterthought until the discovery of RNA editing 30 years ago. -

A Gene-Level Methylome-Wide Association Analysis Identifies Novel
bioRxiv preprint doi: https://doi.org/10.1101/2020.07.13.201376; this version posted July 14, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 A gene-level methylome-wide association analysis identifies novel 2 Alzheimer’s disease genes 1 1 2 3 4 3 Chong Wu , Jonathan Bradley , Yanming Li , Lang Wu , and Hong-Wen Deng 1 4 Department of Statistics, Florida State University; 2 5 Department of Biostatistics & Data Science, University of Kansas Medical Center; 3 6 Population Sciences in the Pacific Program, University of Hawaii Cancer center; 4 7 Tulane Center for Biomedical Informatics and Genomics, Deming Department of Medicine, 8 Tulane University School of Medicine 9 Corresponding to: Chong Wu, Assistant Professor, Department of Statistics, Florida State 10 University, email: [email protected] 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.07.13.201376; this version posted July 14, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 11 Abstract 12 Motivation: Transcriptome-wide association studies (TWAS) have successfully facilitated the dis- 13 covery of novel genetic risk loci for many complex traits, including late-onset Alzheimer’s disease 14 (AD). However, most existing TWAS methods rely only on gene expression and ignore epige- 15 netic modification (i.e., DNA methylation) and functional regulatory information (i.e., enhancer- 16 promoter interactions), both of which contribute significantly to the genetic basis ofAD. -

Mohammad Karbaschi Thesis
STRUCTURAL, PHYSIOLOGICAL AND MOLECULAR CHARACTERISATION OF THE AUSTRALIAN NATIVE RESURRECTION GRASS TRIPOGON LOLIIFORMIS (F.MUELL.) C.E.HUBB. DURING DEHYDRATION AND REHYDRATION Mohammad Reza Karbaschi Submitted in fulfilment of the requirements for the degree of Doctor of Philosophy Centre for Tropical Crops and Biocommodities Science and Engineering Faculty Queensland University of Technology November 2015 Keywords Arabidopsis thaliana; Agrobacterium-mediated transformation; Anatomy; Anti-apoptotic proteins; BAG4; Escherichia coli; Bulliform cells; C4 photosynthesis; Cell wall folding; Cell membrane integrity; Chaperone-mediated autophagy; Chlorophyll fluorescence; Hsc70/Hsp70; Desiccation tolerance, Dehydration; Drought; Electrolyte leakage; Freehand sectioning; Homoiochlorophyllous; Leaf structure; Leaf folding; Reactive oxygen species (ROS); Resurrection plant; Morphology; Monocotyledon; Nicotiana benthamiana; Photosynthesis; Physiology; Plant tissue; Programed cell death (PCD); Propidium iodide staining; Protein microarray chip; Sclerenchymatous tissue; Stress; Structure; Tripogon loliiformis; Ubiquitin; Vacuole fragmentation; Kranz anatomy; XyMS+; Structural, physiological and molecular characterisation of the Australian native resurrection grass Tripogon loliiformis (F.Muell.) C.E.Hubb. during dehydration and rehydration i Abstract Plants, as sessile organisms must continually adapt to environmental changes. Water deficit is one of the major environmental stresses that affects plants. While most plants can tolerate moderate dehydration -
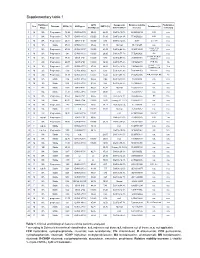
Supplementary Table 2 Supplementary Table 1
Supplementary table 1 Rai/ Binet IGHV Cytogenetic Relative viability Fludarabine- Sex Outcome CD38 (%) IGHV gene ZAP70 (%) Treatment (s) Stage identity (%) abnormalities* increase refractory 1 M 0/A Progressive 14,90 IGHV3-64*05 99,65 28,20 Del17p 18.0% 62,58322819 FCR n.a. 2 F 0/A Progressive 78,77 IGHV3-48*03 100,00 51,90 Del17p 24.8% 77,88052021 FCR n.a. 3 M 0/A Progressive 29,81 IGHV4-b*01 100,00 9,10 Del17p 12.0% 36,48 Len, Chl n.a. 4 M 1/A Stable 97,04 IGHV3-21*01 97,22 18,11 Normal 85,4191657 n.a. n.a. Chl+O, PCR, 5 F 0/A Progressive 87,00 IGHV4-39*07 100,00 43,20 Del13q 68.3% 35,23314039 n.a. HDMP+R 6 M 0/A Progressive 1,81 IGHV3-43*01 100,00 20,90 Del13q 77.7% 57,52490626 Chl n.a. Chl, FR, R-CHOP, 7 M 0/A Progressive 97,80 IGHV1-3*01 100,00 9,80 Del17p 88.5% 48,57389901 n.a. HDMP+R 8 F 2/B Progressive 69,07 IGHV5-a*03 100,00 16,50 Del17p 77.2% 107,9656878 FCR, BA No R-CHOP, FCR, 9 M 1/A Progressive 2,13 IGHV3-23*01 97,22 29,80 Del11q 16.3% 134,5866919 Yes Flavopiridol, BA 10 M 2/A Progressive 0,36 IGHV3-30*02 92,01 0,38 Del13q 81.9% 78,91844953 Unknown n.a. 11 M 2/B Progressive 15,17 IGHV3-20*01 100,00 13,20 Del11q 95.3% 75,52880995 FCR, R-CHOP, BR No 12 M 0/A Stable 0,14 IGHV3-30*02 90,62 7,40 Del13q 13.0% 13,0939004 n.a. -

DULIP: a Dual Luminescence-Based Co-Immunoprecipitation Assay for Interactome Mapping in Mammalian Cells
Repository of the Max Delbrück Center for Molecular Medicine (MDC) in the Helmholtz Association http://edoc.mdc-berlin.de/14998 DULIP: A dual luminescence-based co-immunoprecipitation assay for interactome mapping in mammalian cells Trepte, P., Buntru, A., Klockmeier, K., Willmore, L., Arumughan, A., Secker, C., Zenkner, M., Brusendorf, L., Rau, K., Redel, A., Wanker, E.E. NOTICE: this is the author’s version of a work that was accepted for publication in the Journal of Molecular Biology. Changes resulting from the publishing process, such as peer review, editing, corrections, structural formatting, and other quality control mechanisms may not be reflected in this document. Changes may have been made to this work since it was submitted for publication. A definitive version was subsequently published in: Journal of Molecular Biology 2015 MMM DD ; 427(21): 3375-3388 doi: 10.1016/j.jmb.2015.08.003 Publisher: Elsevier © 2015, Elsevier. This work is licensed under the Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/ or send a letter to Creative Commons, PO Box 1866, Mountain View, CA 94042, USA. DULIP: A DUAL LUMINESCENCE-BASED CO-IMMUNOPRECIPITATION ASSAY FOR INTERACTOME MAPPING IN MAMMALIAN CELLS Philipp Treptea, Alexander Buntrua#, Konrad Klockmeiera#, Lindsay Willmorea, Anup Arumughana, Christopher Seckera, Martina Zenknera, Lydia Brusendorfa, Kirstin Raua, Alexandra Redela and Erich E Wankera* a Neuroproteomics, Max Delbrueck Center for Molecular Medicine, Robert-Roessle- Straße 10, 13125 Berlin, Germany # Contributed equally * Corresponding author, E-mail address: [email protected], Telephone: +49- 30-9406-2157, Fax: +49-30-9406-2552 ABSTRACT Mapping of protein-protein interactions (PPIs) is critical for understanding protein function and complex biological processes. -

Anti-BNIP2 Antibody (ARG58338)
Product datasheet [email protected] ARG58338 Package: 100 μl anti-BNIP2 antibody Store at: -20°C Summary Product Description Rabbit Polyclonal antibody recognizes BNIP2 Tested Reactivity Hu, Ms Tested Application ICC/IF, WB Host Rabbit Clonality Polyclonal Isotype IgG Target Name BNIP2 Antigen Species Human Immunogen Recombinant fusion protein corresponding to aa. 125-280 of Human BNIP2 (NP_004321.2). Conjugation Un-conjugated Alternate Names BNIP-2; BCL2/adenovirus E1B 19 kDa protein-interacting protein 2; NIP2 Application Instructions Application table Application Dilution ICC/IF 1:10 - 1:100 WB 1:500 - 1:2000 Application Note * The dilutions indicate recommended starting dilutions and the optimal dilutions or concentrations should be determined by the scientist. Positive Control SW480 Calculated Mw 36 kDa Observed Size 45-55 kDa Properties Form Liquid Purification Affinity purified. Buffer PBS (pH 7.3), 0.02% Sodium azide and 50% Glycerol. Preservative 0.02% Sodium azide Stabilizer 50% Glycerol Storage instruction For continuous use, store undiluted antibody at 2-8°C for up to a week. For long-term storage, aliquot and store at -20°C. Storage in frost free freezers is not recommended. Avoid repeated freeze/thaw cycles. Suggest spin the vial prior to opening. The antibody solution should be gently mixed before use. www.arigobio.com 1/2 Note For laboratory research only, not for drug, diagnostic or other use. Bioinformation Gene Symbol BNIP2 Gene Full Name BCL2/adenovirus E1B 19kDa interacting protein 2 Background This gene is a member of the BCL2/adenovirus E1B 19 kd-interacting protein (BNIP) family. It interacts with the E1B 19 kDa protein, which protects cells from virally-induced cell death. -

Supplemental Information
Supplemental information Dissection of the genomic structure of the miR-183/96/182 gene. Previously, we showed that the miR-183/96/182 cluster is an intergenic miRNA cluster, located in a ~60-kb interval between the genes encoding nuclear respiratory factor-1 (Nrf1) and ubiquitin-conjugating enzyme E2H (Ube2h) on mouse chr6qA3.3 (1). To start to uncover the genomic structure of the miR- 183/96/182 gene, we first studied genomic features around miR-183/96/182 in the UCSC genome browser (http://genome.UCSC.edu/), and identified two CpG islands 3.4-6.5 kb 5’ of pre-miR-183, the most 5’ miRNA of the cluster (Fig. 1A; Fig. S1 and Seq. S1). A cDNA clone, AK044220, located at 3.2-4.6 kb 5’ to pre-miR-183, encompasses the second CpG island (Fig. 1A; Fig. S1). We hypothesized that this cDNA clone was derived from 5’ exon(s) of the primary transcript of the miR-183/96/182 gene, as CpG islands are often associated with promoters (2). Supporting this hypothesis, multiple expressed sequences detected by gene-trap clones, including clone D016D06 (3, 4), were co-localized with the cDNA clone AK044220 (Fig. 1A; Fig. S1). Clone D016D06, deposited by the German GeneTrap Consortium (GGTC) (http://tikus.gsf.de) (3, 4), was derived from insertion of a retroviral construct, rFlpROSAβgeo in 129S2 ES cells (Fig. 1A and C). The rFlpROSAβgeo construct carries a promoterless reporter gene, the β−geo cassette - an in-frame fusion of the β-galactosidase and neomycin resistance (Neor) gene (5), with a splicing acceptor (SA) immediately upstream, and a polyA signal downstream of the β−geo cassette (Fig. -

The UVB-Induced Gene Expression Profile of Human Epidermis in Vivo Is Different from That of Cultured Keratinocytes
Oncogene (2006) 25, 2601–2614 & 2006 Nature Publishing Group All rights reserved 0950-9232/06 $30.00 www.nature.com/onc ORIGINAL ARTICLE The UVB-induced gene expression profile of human epidermis in vivo is different from that of cultured keratinocytes CD Enk1, J Jacob-Hirsch2, H Gal3, I Verbovetski4, N Amariglio2, D Mevorach4, A Ingber1, D Givol3, G Rechavi2 and M Hochberg1 1Department of Dermatology, The Hadassah-Hebrew University Medical Center, Jerusalem, Israel; 2Department of Pediatric Hemato-Oncology and Functional Genomics, Safra Children’s Hospital, Sheba Medical Center and Sackler School of Medicine, Tel-Aviv University,Tel Aviv, Israel; 3Department of Molecular Cell Biology, Weizmann Institute of Science, Rehovot, Israel and 4The Laboratory for Cellular and Molecular Immunology, Department of Medicine, The Hadassah-Hebrew University Medical Center, Jerusalem, Israel In order to obtain a comprehensive picture of the radiation. UVB, with a wavelength range between 290 molecular events regulating cutaneous photodamage of and 320 nm, represents one of the most important intact human epidermis, suction blister roofs obtained environmental hazards affectinghuman skin (Hahn after a single dose of in vivo ultraviolet (UV)B exposure and Weinberg, 2002). To protect itself against the were used for microarray profiling. We found a changed DNA-damaging effects of sunlight, the skin disposes expression of 619 genes. Half of the UVB-regulated genes over highly complicated cellular programs, including had returned to pre-exposure baseline levels at 72 h, cell-cycle arrest, DNA repair and apoptosis (Brash et al., underscoring the transient character of the molecular 1996). Failure in selected elements of these defensive cutaneous UVB response. -

Study on Apoptosis in Human Deciduous Tooth Pulp Cells Original
Yuko Okai et al.: Apoptosis in Human Deciduous Tooth Pulp Cells Journal of Hard Tissue Biology 21[4] (2012) p413-420 © 2012 The Hard Tissue Biology Network Association Printed in Japan, All rights reserved. CODEN-JHTBFF, ISSN 1341-7649 Original Study on Apoptosis in Human Deciduous Tooth Pulp Cells Yuko Okai1), Kyoko Harada1), Kiyoshi Ohura2) and Kenji Arita1) 1)Department of Pediatric Dentistry, Osaka Dental University,Osaka, Japan 2)Department of Pharmacology, Osaka Dental University, Osaka, Japan (Accepted for publication, July 5, 2012) Abstract: Dental pulp plays an important role in tooth vitality. The pulp of deciduous tooth thus shows vigorous metabolism and repeated, vigorous cell regeneration and proliferation. Furthermore, natural, programmed cell death (apoptosis) frequently occurs as part of this cell regeneration and proliferation process. Apoptosis is defined as cell death governed by intracellular signals (active death of the cell triggered by changes in physiological or external conditions), and the birth of new cells occurs simultaneously with cell death. Apoptosis thus functions as a driving force for morphological and other changes in organisms, and is essential for phenomena such as ontogenetic processes or aging. However, In order to clarify the mechanism of apoptosis in the deciduous tooth pulp cells, we used SuperArray analysis and immunostaining to examine both deciduous and permanent tooth pulp cells. Of the 84 major genes involved in apoptosis, seven showed greater expression in the deciduous tooth pulp cells than in the permanent tooth pulp cells. Gene expression of TP53BP2, CRADD, BAK1, BIRC3, CASP8, CASP2 and TP73 in the deciduous tooth pulp were 16.1, 9.0, 8.5, 8.1, 5.9, 5.8 and 3.2 times greater than those in the permanent tooth pulp cells, respectively. -

Inhibition of Mitochondrial Complex II in Neuronal Cells Triggers Unique
www.nature.com/scientificreports OPEN Inhibition of mitochondrial complex II in neuronal cells triggers unique pathways culminating in autophagy with implications for neurodegeneration Sathyanarayanan Ranganayaki1, Neema Jamshidi2, Mohamad Aiyaz3, Santhosh‑Kumar Rashmi4, Narayanappa Gayathri4, Pulleri Kandi Harsha5, Balasundaram Padmanabhan6 & Muchukunte Mukunda Srinivas Bharath7* Mitochondrial dysfunction and neurodegeneration underlie movement disorders such as Parkinson’s disease, Huntington’s disease and Manganism among others. As a corollary, inhibition of mitochondrial complex I (CI) and complex II (CII) by toxins 1‑methyl‑4‑phenylpyridinium (MPP+) and 3‑nitropropionic acid (3‑NPA) respectively, induced degenerative changes noted in such neurodegenerative diseases. We aimed to unravel the down‑stream pathways associated with CII inhibition and compared with CI inhibition and the Manganese (Mn) neurotoxicity. Genome‑wide transcriptomics of N27 neuronal cells exposed to 3‑NPA, compared with MPP+ and Mn revealed varied transcriptomic profle. Along with mitochondrial and synaptic pathways, Autophagy was the predominant pathway diferentially regulated in the 3‑NPA model with implications for neuronal survival. This pathway was unique to 3‑NPA, as substantiated by in silico modelling of the three toxins. Morphological and biochemical validation of autophagy markers in the cell model of 3‑NPA revealed incomplete autophagy mediated by mechanistic Target of Rapamycin Complex 2 (mTORC2) pathway. Interestingly, Brain Derived Neurotrophic Factor -

Hsa-Mir-21-5P
Supporting Information An exosomal urinary miRNA signature for early diagnosis of renal fibrosis in Lupus Nephritis 1 2 2 1 1 Cristina Solé , Teresa Moliné , Marta Vidal , Josep Ordi-Ros , Josefina Cortés-Hernández 1Hospital Universitari Vall d’Hebron, Vall d’Hebron Research Institute (VHIR), Lupus Unit, Barcelona, Spain; [email protected] (C.S); [email protected] (J.O-R.); [email protected] (J.C-H.) 2 Hospital Universitari Vall d’Hebron, Department of Renal Pathology, Barcelona, Spain; [email protected] (T.M); [email protected] (M.V). INDEX 1. SI Materials and Methods......................................................................................3 2. SI References…………………………………………………………………………….7 2. SI Figures................................................................................................................8 4. SI Tables................................................................................................................21 2 SI Materials and Methods Patients and samples Patients with biopsy-proven active LN were recruited from the Lupus Unit at Vall d’Hebron Hospital (N=45). All patients fulfilled at least 4 of the American College of rheumatology (ACR) revised classification criteria for SLE [1]. Healthy donors were used as controls (N=20). Urine samples were collected from each patient 1 day before renal biopsy and processed immediately to be stored at -80ºC. Patients with urinary tract infection, diabetes mellitus, pregnancy, malignancy and non-lupus-related renal failure were excluded. In addition, key laboratory measurements were obtained including complement levels (C3 and C4), anti-double-stranded DNA (anti-dsDNA), 24-h proteinuria, blood urea nitrogen (BUN), serum creatinine and the estimated glomerular filtration ratio (eGFR) using the Chronic Kidney Disease Epidemiology Collaboration (CKD-EPI) formula [2]. SLE disease activity was assessed by the SLE Disease Activity Index 2000 update (SLEDAI-2Ks; range 0–105) [3].