A Gene-Level Methylome-Wide Association Analysis Identifies Novel
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Seq2pathway Vignette
seq2pathway Vignette Bin Wang, Xinan Holly Yang, Arjun Kinstlick May 19, 2021 Contents 1 Abstract 1 2 Package Installation 2 3 runseq2pathway 2 4 Two main functions 3 4.1 seq2gene . .3 4.1.1 seq2gene flowchart . .3 4.1.2 runseq2gene inputs/parameters . .5 4.1.3 runseq2gene outputs . .8 4.2 gene2pathway . 10 4.2.1 gene2pathway flowchart . 11 4.2.2 gene2pathway test inputs/parameters . 11 4.2.3 gene2pathway test outputs . 12 5 Examples 13 5.1 ChIP-seq data analysis . 13 5.1.1 Map ChIP-seq enriched peaks to genes using runseq2gene .................... 13 5.1.2 Discover enriched GO terms using gene2pathway_test with gene scores . 15 5.1.3 Discover enriched GO terms using Fisher's Exact test without gene scores . 17 5.1.4 Add description for genes . 20 5.2 RNA-seq data analysis . 20 6 R environment session 23 1 Abstract Seq2pathway is a novel computational tool to analyze functional gene-sets (including signaling pathways) using variable next-generation sequencing data[1]. Integral to this tool are the \seq2gene" and \gene2pathway" components in series that infer a quantitative pathway-level profile for each sample. The seq2gene function assigns phenotype-associated significance of genomic regions to gene-level scores, where the significance could be p-values of SNPs or point mutations, protein-binding affinity, or transcriptional expression level. The seq2gene function has the feasibility to assign non-exon regions to a range of neighboring genes besides the nearest one, thus facilitating the study of functional non-coding elements[2]. Then the gene2pathway summarizes gene-level measurements to pathway-level scores, comparing the quantity of significance for gene members within a pathway with those outside a pathway. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Dynamic Transcriptomic Profiles of Zebrafish Gills in Response to Zinc
Zheng et al. BMC Genomics 2010, 11:548 http://www.biomedcentral.com/1471-2164/11/548 RESEARCH ARTICLE Open Access Dynamic transcriptomic profiles of zebrafish gills in response to zinc depletion Dongling Zheng1,4, Peter Kille2, Graham P Feeney2, Phil Cunningham1, Richard D Handy3, Christer Hogstrand1* Abstract Background: Zinc deficiency is detrimental to organisms, highlighting its role as an essential micronutrient contributing to numerous biological processes. To investigate the underlying molecular events invoked by zinc depletion we performed a temporal analysis of transcriptome changes observed within the zebrafish gill. This tissue represents a model system for studying ion absorption across polarised epithelial cells as it provides a major pathway for fish to acquire zinc directly from water whilst sharing a conserved zinc transporting system with mammals. Results: Zebrafish were treated with either zinc-depleted (water = 2.61 μgL-1; diet = 26 mg kg-1) or zinc-adequate (water = 16.3 μgL-1; diet = 233 mg kg-1) conditions for two weeks. Gill samples were collected at five time points and transcriptome changes analysed in quintuplicate using a 16K oligonucleotide array. Of the genes represented the expression of a total of 333 transcripts showed differential regulation by zinc depletion (having a fold-change greater than 1.8 and an adjusted P-value less than 0.1, controlling for a 10% False Discovery Rate). Down-regulation was dominant at most time points and distinct sets of genes were regulated at different stages. Annotation enrichment analysis revealed that ‘Developmental Process’ was the most significantly overrepresented Biological Process GO term (P = 0.0006), involving 26% of all regulated genes. -

The Interactome of KRAB Zinc Finger Proteins Reveals the Evolutionary History of Their Functional Diversification
Resource The interactome of KRAB zinc finger proteins reveals the evolutionary history of their functional diversification Pierre-Yves Helleboid1,†, Moritz Heusel2,†, Julien Duc1, Cécile Piot1, Christian W Thorball1, Andrea Coluccio1, Julien Pontis1, Michaël Imbeault1, Priscilla Turelli1, Ruedi Aebersold2,3,* & Didier Trono1,** Abstract years ago (MYA) (Imbeault et al, 2017). Their products harbor an N-terminal KRAB (Kru¨ppel-associated box) domain related to that of Krüppel-associated box (KRAB)-containing zinc finger proteins Meisetz (a.k.a. PRDM9), a protein that originated prior to the diver- (KZFPs) are encoded in the hundreds by the genomes of higher gence of chordates and echinoderms, and a C-terminal array of zinc vertebrates, and many act with the heterochromatin-inducing fingers (ZNF) with sequence-specific DNA-binding potential (Urru- KAP1 as repressors of transposable elements (TEs) during early tia, 2003; Birtle & Ponting, 2006; Imbeault et al, 2017). KZFP genes embryogenesis. Yet, their widespread expression in adult tissues multiplied by gene and segment duplication to count today more and enrichment at other genetic loci indicate additional roles. than 350 and 700 representatives in the human and mouse Here, we characterized the protein interactome of 101 of the ~350 genomes, respectively (Urrutia, 2003; Kauzlaric et al, 2017). A human KZFPs. Consistent with their targeting of TEs, most KZFPs majority of human KZFPs including all primate-restricted family conserved up to placental mammals essentially recruit KAP1 and members target sequences derived from TEs, that is, DNA trans- associated effectors. In contrast, a subset of more ancient KZFPs posons, ERVs (endogenous retroviruses), LINEs, SINEs (long and rather interacts with factors related to functions such as genome short interspersed nuclear elements, respectively), or SVAs (SINE- architecture or RNA processing. -

DULIP: a Dual Luminescence-Based Co-Immunoprecipitation Assay for Interactome Mapping in Mammalian Cells
Repository of the Max Delbrück Center for Molecular Medicine (MDC) in the Helmholtz Association http://edoc.mdc-berlin.de/14998 DULIP: A dual luminescence-based co-immunoprecipitation assay for interactome mapping in mammalian cells Trepte, P., Buntru, A., Klockmeier, K., Willmore, L., Arumughan, A., Secker, C., Zenkner, M., Brusendorf, L., Rau, K., Redel, A., Wanker, E.E. NOTICE: this is the author’s version of a work that was accepted for publication in the Journal of Molecular Biology. Changes resulting from the publishing process, such as peer review, editing, corrections, structural formatting, and other quality control mechanisms may not be reflected in this document. Changes may have been made to this work since it was submitted for publication. A definitive version was subsequently published in: Journal of Molecular Biology 2015 MMM DD ; 427(21): 3375-3388 doi: 10.1016/j.jmb.2015.08.003 Publisher: Elsevier © 2015, Elsevier. This work is licensed under the Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/ or send a letter to Creative Commons, PO Box 1866, Mountain View, CA 94042, USA. DULIP: A DUAL LUMINESCENCE-BASED CO-IMMUNOPRECIPITATION ASSAY FOR INTERACTOME MAPPING IN MAMMALIAN CELLS Philipp Treptea, Alexander Buntrua#, Konrad Klockmeiera#, Lindsay Willmorea, Anup Arumughana, Christopher Seckera, Martina Zenknera, Lydia Brusendorfa, Kirstin Raua, Alexandra Redela and Erich E Wankera* a Neuroproteomics, Max Delbrueck Center for Molecular Medicine, Robert-Roessle- Straße 10, 13125 Berlin, Germany # Contributed equally * Corresponding author, E-mail address: [email protected], Telephone: +49- 30-9406-2157, Fax: +49-30-9406-2552 ABSTRACT Mapping of protein-protein interactions (PPIs) is critical for understanding protein function and complex biological processes. -

Anti-BNIP2 Antibody (ARG58338)
Product datasheet [email protected] ARG58338 Package: 100 μl anti-BNIP2 antibody Store at: -20°C Summary Product Description Rabbit Polyclonal antibody recognizes BNIP2 Tested Reactivity Hu, Ms Tested Application ICC/IF, WB Host Rabbit Clonality Polyclonal Isotype IgG Target Name BNIP2 Antigen Species Human Immunogen Recombinant fusion protein corresponding to aa. 125-280 of Human BNIP2 (NP_004321.2). Conjugation Un-conjugated Alternate Names BNIP-2; BCL2/adenovirus E1B 19 kDa protein-interacting protein 2; NIP2 Application Instructions Application table Application Dilution ICC/IF 1:10 - 1:100 WB 1:500 - 1:2000 Application Note * The dilutions indicate recommended starting dilutions and the optimal dilutions or concentrations should be determined by the scientist. Positive Control SW480 Calculated Mw 36 kDa Observed Size 45-55 kDa Properties Form Liquid Purification Affinity purified. Buffer PBS (pH 7.3), 0.02% Sodium azide and 50% Glycerol. Preservative 0.02% Sodium azide Stabilizer 50% Glycerol Storage instruction For continuous use, store undiluted antibody at 2-8°C for up to a week. For long-term storage, aliquot and store at -20°C. Storage in frost free freezers is not recommended. Avoid repeated freeze/thaw cycles. Suggest spin the vial prior to opening. The antibody solution should be gently mixed before use. www.arigobio.com 1/2 Note For laboratory research only, not for drug, diagnostic or other use. Bioinformation Gene Symbol BNIP2 Gene Full Name BCL2/adenovirus E1B 19kDa interacting protein 2 Background This gene is a member of the BCL2/adenovirus E1B 19 kd-interacting protein (BNIP) family. It interacts with the E1B 19 kDa protein, which protects cells from virally-induced cell death. -

Genome-Wide Association Study in Collaborative Cross Mice Revealed a Skeletal
bioRxiv preprint doi: https://doi.org/10.1101/094698; this version posted December 16, 2016. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC 4.0 International license. Title Genome-wide association study in Collaborative Cross mice revealed a skeletal role for Rhbdf2 Authors Roei Levy1,2, Clemence Levet3, Keren Cohen1, Matthew Freeman3, Richard Mott4, Fuad Iraqi5, Yankel Gabet1 Affiliations 1 Department of Anatomy and Anthropology, 2 Department of Human Molecular Genetics and Biochemistry, and 5 Department of Clinical Microbiology and Immunology, Sackler Medical School, Tel Aviv University. 3 Dunn School of Pathology, South Parks Road, Oxford OX1 3RE. 4 UCL Genetics Institute, University College London, Gower St., London, WC1E 6BT, UK. Abstract A growing concern that overshadows the increased life expectancy developed countries have been witnessing during the last decades is an accompanying bone loss, which often manifests as osteoporosis. Despite ongoing efforts in utilizing genomic data to fully map the genes responsible for bone remodeling, a detailed picture remains to be desired. Here we took advantage of the phenotypic and genetic diversity innate in Collaborative Cross (CC) mice to identify genetic variants associated with microstructural bone characteristics. 1 bioRxiv preprint doi: https://doi.org/10.1101/094698; this version posted December 16, 2016. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. -

Supplemental Information
Supplemental information Dissection of the genomic structure of the miR-183/96/182 gene. Previously, we showed that the miR-183/96/182 cluster is an intergenic miRNA cluster, located in a ~60-kb interval between the genes encoding nuclear respiratory factor-1 (Nrf1) and ubiquitin-conjugating enzyme E2H (Ube2h) on mouse chr6qA3.3 (1). To start to uncover the genomic structure of the miR- 183/96/182 gene, we first studied genomic features around miR-183/96/182 in the UCSC genome browser (http://genome.UCSC.edu/), and identified two CpG islands 3.4-6.5 kb 5’ of pre-miR-183, the most 5’ miRNA of the cluster (Fig. 1A; Fig. S1 and Seq. S1). A cDNA clone, AK044220, located at 3.2-4.6 kb 5’ to pre-miR-183, encompasses the second CpG island (Fig. 1A; Fig. S1). We hypothesized that this cDNA clone was derived from 5’ exon(s) of the primary transcript of the miR-183/96/182 gene, as CpG islands are often associated with promoters (2). Supporting this hypothesis, multiple expressed sequences detected by gene-trap clones, including clone D016D06 (3, 4), were co-localized with the cDNA clone AK044220 (Fig. 1A; Fig. S1). Clone D016D06, deposited by the German GeneTrap Consortium (GGTC) (http://tikus.gsf.de) (3, 4), was derived from insertion of a retroviral construct, rFlpROSAβgeo in 129S2 ES cells (Fig. 1A and C). The rFlpROSAβgeo construct carries a promoterless reporter gene, the β−geo cassette - an in-frame fusion of the β-galactosidase and neomycin resistance (Neor) gene (5), with a splicing acceptor (SA) immediately upstream, and a polyA signal downstream of the β−geo cassette (Fig. -

Inhibition of Mitochondrial Complex II in Neuronal Cells Triggers Unique
www.nature.com/scientificreports OPEN Inhibition of mitochondrial complex II in neuronal cells triggers unique pathways culminating in autophagy with implications for neurodegeneration Sathyanarayanan Ranganayaki1, Neema Jamshidi2, Mohamad Aiyaz3, Santhosh‑Kumar Rashmi4, Narayanappa Gayathri4, Pulleri Kandi Harsha5, Balasundaram Padmanabhan6 & Muchukunte Mukunda Srinivas Bharath7* Mitochondrial dysfunction and neurodegeneration underlie movement disorders such as Parkinson’s disease, Huntington’s disease and Manganism among others. As a corollary, inhibition of mitochondrial complex I (CI) and complex II (CII) by toxins 1‑methyl‑4‑phenylpyridinium (MPP+) and 3‑nitropropionic acid (3‑NPA) respectively, induced degenerative changes noted in such neurodegenerative diseases. We aimed to unravel the down‑stream pathways associated with CII inhibition and compared with CI inhibition and the Manganese (Mn) neurotoxicity. Genome‑wide transcriptomics of N27 neuronal cells exposed to 3‑NPA, compared with MPP+ and Mn revealed varied transcriptomic profle. Along with mitochondrial and synaptic pathways, Autophagy was the predominant pathway diferentially regulated in the 3‑NPA model with implications for neuronal survival. This pathway was unique to 3‑NPA, as substantiated by in silico modelling of the three toxins. Morphological and biochemical validation of autophagy markers in the cell model of 3‑NPA revealed incomplete autophagy mediated by mechanistic Target of Rapamycin Complex 2 (mTORC2) pathway. Interestingly, Brain Derived Neurotrophic Factor -
Download Validation Data
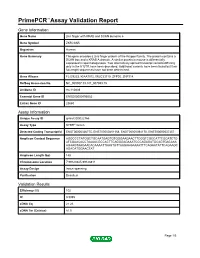
PrimePCR™Assay Validation Report Gene Information Gene Name zinc finger with KRAB and SCAN domains 5 Gene Symbol ZKSCAN5 Organism Human Gene Summary This gene encodes a zinc finger protein of the Kruppel family. The protein contains a SCAN box and a KRAB A domain. A similar protein in mouse is differentially expressed in spermatogenesis. Two alternatively spliced transcript variants differing only in the 5' UTR have been described. Additional variants have been found but their full-length sequences have not been determined. Gene Aliases FLJ39233, KIAA1015, MGC33710, ZFP95, ZNF914 RefSeq Accession No. NC_000007.13, NT_007933.15 UniGene ID Hs.110839 Ensembl Gene ID ENSG00000196652 Entrez Gene ID 23660 Assay Information Unique Assay ID qHsaCID0022766 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000326775, ENST00000451158, ENST00000394170, ENST00000537357 Amplicon Context Sequence AGGCCCTATGGCTGCAATGAGTGTGGGAAGAACTTCGGTCGCCATTCGCATCTG ATCGAACACCTAAAACGCCACTTCAGGGAGAAATCCCAGAGATGCAGTGACAAA AGAAGTAAGAACACAAAATTAAGTGTTAAGAAGAAAATTTCAGAATATTCAGAAGC AGACATGGAACTAT Amplicon Length (bp) 148 Chromosome Location 7:99123945-99128811 Assay Design Intron-spanning Purification Desalted Validation Results Efficiency (%) 102 R2 0.9985 cDNA Cq 21.26 cDNA Tm (Celsius) 81.5 Page 1/5 PrimePCR™Assay Validation Report gDNA Cq 34.61 Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 2/5 PrimePCR™Assay Validation Report ZKSCAN5, Human Amplification Plot Amplification of cDNA generated from -

Genetic Deletion of Amphiregulin Restores the Normal Skin Phenotype in a Mouse Model of the Human Skin Disease Tylosis Vishnu Hosur*, Benjamin E
© 2017. Published by The Company of Biologists Ltd | Biology Open (2017) 6, 1174-1179 doi:10.1242/bio.026260 RESEARCH ARTICLE Genetic deletion of amphiregulin restores the normal skin phenotype in a mouse model of the human skin disease tylosis Vishnu Hosur*, Benjamin E. Low, Leonard D. Shultz and Michael V. Wiles ABSTRACT mutations in human RHBDF2 lead to enhanced AREG secretion In humans, gain-of-function (GOF) mutations in RHBDF2 cause the and tylosis. We provide evidence that Rhbdf2 GOF mutations skin disease tylosis. We generated a mouse model of human tylosis enhance AREG secretion to cause hyperplasia and hyperkeratosis. and show that GOF mutations in RHBDF2 cause tylosis by enhancing In addition, we show that genetic ablation of AREG attenuates skin P159L/P159L the amount of amphiregulin (AREG) secretion. Furthermore, we show disease in the Rhbdf2 mouse model of human tylosis. that genetic disruption of AREG ameliorates skin pathology in mice Together, our data strongly suggest that inhibition of AREG could carrying the human tylosis disease mutation. Collectively, our data have potential therapeutic value in the treatment of tylosis. suggest that RHBDF2 plays a critical role in regulating EGFR signaling and its downstream events, including development of RESULTS AND DISCUSSION tylosis, by facilitating enhanced secretion of AREG. Thus, targeting The human tylosis disease mutation enhances AREG AREG could have therapeutic benefit in the treatment of tylosis. secretion Autosomal dominant mutations in the human RHBDF2 gene cause KEY WORDS: CRISPR/Cas9, EGFR, RHBDF2, Amphiregulin, Tylosis tylosis (Blaydon et al., 2012; Saarinen et al., 2012). Substantial evidence implicates the involvement of AREG-induced constitutive INTRODUCTION activation of the EGFR pathway (Blaydon et al., 2012; Brooke et al., The role of RHBDF2 in enhancing amphiregulin (AREG) secretion, 2014; Hosur et al., 2014) in human tylosis. -

Human Induced Pluripotent Stem Cell–Derived Podocytes Mature Into Vascularized Glomeruli Upon Experimental Transplantation
BASIC RESEARCH www.jasn.org Human Induced Pluripotent Stem Cell–Derived Podocytes Mature into Vascularized Glomeruli upon Experimental Transplantation † Sazia Sharmin,* Atsuhiro Taguchi,* Yusuke Kaku,* Yasuhiro Yoshimura,* Tomoko Ohmori,* ‡ † ‡ Tetsushi Sakuma, Masashi Mukoyama, Takashi Yamamoto, Hidetake Kurihara,§ and | Ryuichi Nishinakamura* *Department of Kidney Development, Institute of Molecular Embryology and Genetics, and †Department of Nephrology, Faculty of Life Sciences, Kumamoto University, Kumamoto, Japan; ‡Department of Mathematical and Life Sciences, Graduate School of Science, Hiroshima University, Hiroshima, Japan; §Division of Anatomy, Juntendo University School of Medicine, Tokyo, Japan; and |Japan Science and Technology Agency, CREST, Kumamoto, Japan ABSTRACT Glomerular podocytes express proteins, such as nephrin, that constitute the slit diaphragm, thereby contributing to the filtration process in the kidney. Glomerular development has been analyzed mainly in mice, whereas analysis of human kidney development has been minimal because of limited access to embryonic kidneys. We previously reported the induction of three-dimensional primordial glomeruli from human induced pluripotent stem (iPS) cells. Here, using transcription activator–like effector nuclease-mediated homologous recombination, we generated human iPS cell lines that express green fluorescent protein (GFP) in the NPHS1 locus, which encodes nephrin, and we show that GFP expression facilitated accurate visualization of nephrin-positive podocyte formation in