Structure–Activity Relationships of Benzothiazole-Based Hsp90 C
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

060528 Tramnetz.Pdf
h Fahrscheine bitte vor Fahrtantritt entwerten. Legende Service MetroTram + Straßenbahn-Netz Tarifbereich Berlin A B C A B Haltestellen in Berlin C Haltestellen in Brandenburg Tickets must be validated before use. 2 S+U-Bahn-Linie mit AB 7 Umsteigemöglichkeit Berliner Verkehrsbetriebe (BVG) S+U-Bahn + MetroBus-Netz M1 MetroTram-Linie 10773 Berlin 50 Straßenbahn-Linie www.bvg.de - [email protected] imode.bvg.de - wap.bvg.de M19 MetroBus-Linie S Waidmannslust iT M1 Rosenthal Nord B4 50 Guyotstr. < BVG Call Center: (030) 19 44 9 Halt nur in Pfeilrichtung Hauptstr./Friedrich-Engels-Str. Hugenottenplatz Tag und Nacht. Rund um die Uhr. 6 Arnouxstr. f0 b0 Bus-Anbindung zum Flughafen Auch am Wochenende. Wiesenwinkel 8 M21 S+U Wittenau Uhlandstr./ B4 Navarraplatz Fernbahnhof BVG Fahrinfo SMS S Tegel S+U Karl-Bonhoeffer-Nervenklinik Wilhelmsruher Damm Angerweg M1 Schillerstr. Französisch Buchholz Kirche p BVG-Fundbüro Standort-Nr. senden an 77 3 77 S8 Berlin Blankenfelder Str. Nordendstr. Waldemarstr. BVG Fundbüro Uhlandstr. Nordend Rosenthaler Str. Barrierefreier Nahverkehr am U-Bhf Kleistpark/U-Bhf Bülowstr. Potsdamer Str. 182 (nahe Pallasstraße) S Schönholz Platanenstr. Marienstr./Pasewalker Str. uT S Wartenberg B4 B4 MetroTram/Tram Heinrich-Böll-Str. 10783 Berlin (Schöneberg) U6 Skladanowskystr. Kuckhoffstr. Pasewalker Str./Blankenburger Weg bedingt barrierefreie Fahrzeuge u4 (030) 19 44 9 M21 Pastor-Niemöller-Platz U8 H.-Hesse-Str./ Pankower Str. Grabbeallee/Pastor-Niemöller-Platz Waldstr. Galenusstr. S1 S8 S85 B4 Tschaikowskistr. M2 Heinersdorf B4 Zingster Str. M4 M5 StiftswegMendelstr. S Pankow-Heinersdorf M4 M17 Falkenberg B4 S-Bahn Berlin GmbH Bürgerpark Pankow www.s-bahn-berlin.de U Kurt-Schumacher- Würtzstr. -

Ich Fahre Jetzt Viel Mit Der BVG« INTRO
Nr. 9 / November 2020 DAS MAGAZIN DER BERLINER VERKEHRSBETRIEBE Land und Stadt Mit dem Bus 110 von Dahlem nach Halensee INTERVIEW MIT SCHAUSPIELERIN KAROLINE SCHUCH »Ich fahre jetzt viel mit der BVG« INTRO INHALT 6 Karoline Schuch Stationen meines Lebens GAN(S)ZUnsere KÖSTLICH TagesfahrtenFRÖHLICHE LANDPARTIE BONOVO-SPEZIAL Im Interview erzählt die Schauspielerin, warum AUF DEM SPARGELHOF ZUR WINTERZEIT sie als TV-Kommissarin auch im Knast war und Leistungen Leistungen noch heute gerne U-Bahn fährt + Komfortbus mit Bordservice + Komfortbus mit Bordservice + Bonovo-Reiseleitung + Bonovo-Reiseleitung + 3-Gang-Menü mit Gänsekeule + Mittagessen (Ersatzgericht möglich) + 1x Kaffee + Eintritt Museum Neuruppin + 1x Sekt Martinsganszeit TAGESFAHRT in Kremmen TAGESFAHRT Winterlandpartie 12.11.20 € 66,-p.P. 17.11.20 € 47,-p.P. 12 Mahlzeit! AUF DEN SPUREN DES VORFREUDE AUF BVG verbindet Berlin SCHWARZEN ABTES WEIHNACHTEN Wie die Lebensmittelretter Leistungen BEIM ENTENBÜFFET! Leckereien mit aussortier- 14 Weide und Wiese + Komfortbus mit Bordservice Leistungen + Bonovo-Reiseleitung: ten Zutaten bereiten Eine Linie, zwei Welten + Komfortbus mit Bordservice Frau Pawlak + Bonovo-Reiseleitung Mit dem 110er Bus von der Domäne + Mittagessen + Entenbüffet wie beschrieben + Führung Klostergelände Dahlem zum Hochmeisterplatz + Kaffeegedeck + Führung/Verkostung Klosterbrauerei Schlaubetal & 4 Was war / Was kommt 9 Der Monat mit der BVG EDITORIAL TAGESFAHRT Klosterbrauerei Neuzelle TAGESFAHRT Klaistow p.P. p.P. 10 Hinter den Kulissen 20.11.20 24.11.20 € 71,- € 64,- Stefan Büttner kontrolliert die Liebe Leser*innen, Gleisanlagen der BVG LICHTERFAHRT MIT KAFFEETRINKEN AUF DEM FERNSEHTURM 18 Tipps vom tip fällt Ihnen etwas auf? Richtig, 20 Im Bahnhof erstmals benutze ich den Leistungen Sonstiges Neues Buch über die U5 + Komfortbus mit Bordservice • Die Mitnahme von Rol- Genderstern in der Ansprache. -
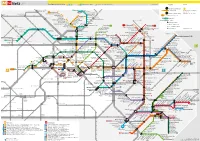
4M4t Netz0s 4U
Netz Tarifbereich Berlin A B C A B Haltestellen in Berlin C Haltestellen in Brandenburg www.BVG.de Legende Service 4M 4t 0s 4u 24 Linie im 24-Stunden-Betrieb Waidmannslust :hE 50 M1 Guyotstr. Arnouxstr. S8 MetroTram täglich Berliner Verkehrsbetriebe (BVG) Wittenau :8 Navarraplatz M1 Hugenotten- S2 www.BVG.de Rosenthal Nord Französisch Buchholz Kirche platz 20 BVG Call Center: 030 19 44 9 Karl-Bonhoeffer- Hauptstr./Friedrich-Engels-Str. Linie im 20-Stunden-Betrieb Tegel Blankenfelder Str. Nervenklinik Wiesenwinkel Rosenthaler Str. M1 MetroTram-Abschnitte Schillerstr. M1 S25 Angerweg Marienstr./Pasewalker Str. Schönholz Waldemarstr. Nordendstr. Blankenburg 50 Straßenbahn Friedrich-Engels-Str./Eichenstr. Nordend Pasewalker Str./Blankenburger Weg Heinrich-Böll-Str. Wartenberg :gE 2 7 S+U-Bahn U6 Am Iderfenngraben Pankower Str. Kuckhoffstr. Umsteigemöglichkeit U8 Pastor-Niemöller-Platz H.-Hesse-Str./ . S1 S2 eg Galenusstr. Grabbeallee/Pastor-Niemöller-Platz Waldstr. > Halt nur in Pfeilrichtung 5 S8 Tschaikowskistr. S Pankow-Heinersdorf M4 M5 Zingster Str. 0A 128 StiftswMendelstr Heinersdorf M2 Falkenberg M4 M17 0b 5 Bürgerpark Pankow Fern- und Regionalbahnhof Kurt-Schumacher-Platz Würtzstr. Rothenbachstr. Zingster Str./Ribnitzer Str. Welsestr. :9U Osloer Str. Drontheimer Str. Rathaus Pankow Pankow Heinersdorf Kirche Falkenberger Ch./Prendener Str. Regionalbahnhof Stand: 3. April 2016 Kirche Ahrenshooper Str. Louise-Schroeder-Platz Am Wasserturm S Hohenschönhausen Osloer Str./ Prerower Platz Prinzenallee :9 :2 Tino-Schwierzina-Str. Osram-Höfe S+U Pankow Hansastr./Malchower Weg Rüdickenstr. 8 0 Am Steinberg M2 12 5 S2 S8 S9 M4 Arnimstr. :7 Ahrensfelde 3 M1 Masurenstr. Feldtmannstr. M1 3 Prenzlauer Promenade/ 5 Stadion Buschallee/Hansastr. 0 Grüntaler Str. Am Steinberg 12 27 Pasedagplatz U Seestr. -

CAFR Includes the Dishiet's Basic Financial Statement Prepared in Accordance with Governrnental Acc'ounting Standards Board Statement 34
SCHOOL DISTRICT OF BERLIN BOROUGH Berlin Borough Board of Education Berlin, New Jersey Comprehensive Annual Financial Report For the Fiscal Year Ended June 30, 2018 Comprehensive Annual Financial Report of the Berlin Borough Board of Education Berlin, New Jersey For the Fiscal Year Ended June 30, 2018 Prepared by Berlin Borough Board of Education Finance Department BERLIN BOROUGH SCHOOL DISTRICT INTRODUCTORY SECTION Page Letter of Transmittal 2 Organizational Chart 7 Roster of Officials 8 Consultants and Advisors 9 FINANCIAL SECTION Independent Auditor's Report 11 K-1 Report on Compliance and on Internal Control Over Financial Reporting Based on an Audit of Financial Statements Performed in Accordance with Government Auditing Standards 14 Required Supplementary Information - Part I Management's Discussion and Analysis 17 Basic Financial Statements A. District-wide Financial Statements: A-1 Statement of Net Position 27 A-2 Statement of Activities 28 B. Fund Financial Statements: Governmental Funds: B-1 Balance Sheet 30 B-2 Statement of Revenues, Expenditures, and Changes in Fund Balances 31 B-3 Reconciliation of the Statement of Revenues, Expenditures, and Changes in Fund Balances of Governmental Funds to the Statement of Activities 32 Proprietary Funds: B-4 Statement of Net Position 33 B-5 Statement of Revenues, Expenses, and Changes in Fund Net Position 34 B-6 Statement of Cash Flows 35 Fiduciary Funds: B-7 Statement of Fiduciary Net Position 36 B-8 Statement of Changes in Fiduciary Net Position 37 Notes to the Financial Statements 38 Page Required Supplementary Information - Part II C. Budgetary Comparison Schedules C-1 Budgetary Comparison Schedule - General Fund 72 C-1a Combining Schedule of Revenues, Expenditures and Changes in Fund Balance - Budget and Actual (if applicable) N/A C-2 Budgetary Comparison Schedule - Special Revenue Fund 78 Notes to the Required Supplementary Information C-3 Budget-to-GAAP Reconciliation 79 Required Supplementary Information - Part III L. -
Author Index
Neuropsychopharmacology (2011) 36, S450–S482 & 2011 American College of Neuropsychopharmacology. All rights reserved 0893-133X/11 S450 www.neuropsychopharmacology.org Author Index A Alcantara, Lyonna F. S431 Alda, Martin . S391 Aarde, Shawn A. S441 Alegria, Dylan . S398 Abazyan, Bagrat. S214 Alexander, Kathleen S . S209 Abazyan, Sofya . S214 Alexander-Bloch, Aaron F . S272 Abbott, Chris. S386 Al-Hasani, Ream . S440 Abbs, Brandon. S314 Alia-Klein, Nelly . S376 Abdallah, Chadi G . S139 Alim, Tanya. S304 Abdel-Hamid, Mona. .S135 Allard, Carolyn B. S139 Abel, Ted . S223 Allen, John . S185, S186, S394 Abelson, James . S144, S406 Allen, Katherine. S134 Abend, Rany . S110 Allen, Wade M . .S171 Abercrombie, Heather C. S158 Almeida, Jorge RC . S290 Aberdam, Daniel . S218 Almli, Lynn M. S256 Abi-Dargham, Anissa . .S20, S282 Almog, Orna . S429 Abrams, Debra J . S129 Altshuler, Lori . S116, S248, S396 Abulseoud, Osama A . S343 Aluisio, Leah . .S191 Aceti, Max. S84 Alvarez-Estrada, Miguel . S179 Acheson, Shawn K . S306 Aman, Michael G. S228 Achtyes, Eric . S232 Amar, Shirly . S414 Adams, David H . .S132 Amaral, David . S31 Adams, Matthew . .S331 Amir, Nader . S143 Addington, Jean. .S10, S240 Amita, Hidetoshi . S447 Adelekun, Adesewa E . S436 Amy, Easton . S182 Adleman, Nancy E . S386 Anand, Amit . .S153 Adler, Caleb M . S159, S297 Anand, Reena . S391 Adler, Lenard . S230 Anastasio, Noelle . S19, S310 Adli, Mazda. .S161 Anders, Martin . S167 Agam, Galila . .S85, S414, S429 Andersen, Susan . .S50, S439 Agarwal, Vishesh . S167 Anderson, Adam . .S171 Aghajanian, George . S62 Anderson, Colleen . .S353 Agid, Ofer . S103 Anderson, George . S198 Agster, Kara L . S83 Anderson, Jeffery . .S153 Ahmari, Susanne E . S75 Anderson, Stewart J . .S115 Ahmed, Saeeduddin . -
Rediscover the Metropolis Berlin Route of Industrial Heritage I Berlin Route of Industrial Heritage
EN Rediscover the Metropolis Berlin Route of Industrial Heritage i Berlin Route of Industrial Heritage Berlin the Electropolis The Berlin Route of Industrial Heritage With Prussia’s first railroad, one of the first three-phase electric The sites and related locations on the Berlin Route of Industrial power plants in Europe, and the first electric streetcar line Heritage are landmarks of the city’s technological, economic, in the world, Berlin was a hub of international economic, tech- and social history. One new site is the royal porcelain manu- nological, and architectural transformation. Beginning in the factory Königliche Porzellan-Manufaktur Berlin (KPM), once a 1880s, this development was driven especially by the electrical model of early industrialization. Today it continues to operate industry. Berlin was a center not only of production but also of at its historic location, thus making it a special symbol of research and development, and it served as a testing ground for continuity and change. In many cases, citizen initiatives are new technologies. Its power, water, and transportation systems to thank for preserving important monuments of Berlin’s set the standard around the world. Around 1900, Berlin was industrial heritage. Visitors are sure to meet knowledgeable for a time the largest metropolis in continental Europe. The enthusiasts there whom they can engage in conversation. cityscape and society underwent rapid changes. Some sites can only be visited as part of a guided tour or by making an appointment ahead of time. The information in Berlin is Industrial Heritage this brochure is subject to change. Please confirm it before making your visit! Old factory buildings, industrial sites, and substations – many of them now repurposed – are an essential component of Berlin’s More information and tips on Berlin’s industrial heritage can unique flair. -

Berlin Metro Map by Zuti
Hohen Mühlenbeck Bernau Borgsdorf Neuendorf Bergfelde Schönfließ Mönchmühle Karow Röntgental Friedenstal Oranienburg Bernau Lehnitz Birkenwerder Hugenotten Navarrapl Buch Zepernick Guyotstr bei Bernau Rosenthal Nord Arnoux HAVEL Französisch Hauptstr Buchholz Kirche Frohnau Friedrich Engels 50 HAVEL Wiesenwinkel Blankenfelder Berlin Angerweg © Copyright Visual IT Ltd Nordendstr Rosenthaler ® Zuti and the Zuti logo are registered trademarks Hermsdorf www.zuti.co.uk Nordend Schillerstr Marienstr BERLIN WALL BERLIN Uhlandstr Pasewalker Blankenburg Hennigsdorf Waldemar Waidmannslust Pasewalker Platanenstr Heinrich Böll Blankenburger Weg Heiligensee Pankower Am Iderfenngraben Kuckhoffstr Pastor Niemöller Platz Schulzendorf Galenusstr Wittenau Hermann Hesse Grabbeallee Waldstr Pastor Niemöller Ahrensfelde REINICKENDORF Ahrensfelde Tschaikowskistr HAVEL Rathaus Würtzstr Wartenberg Reinickendorf Mendelstr Tegel Wilhelmsruh M1 Pankow Zingster Falkenberger Karl B Heinersdorf Prendener Welsestr Nerven Bürgerpark Stiftsweg Heinersdorf Falkenberg Barnimplatz Alt Tegel klinik Alt Reinickendorf Pankow Rathaus Zingster Ribnitzer Schönholz Pankow PANKOW Hohenschönhausen Eichborn Ahrenshooper Niemegker Borsigwerke damm Pankow Rothenbachstr Paracelsus Bad Kirche Prerower U8 Mehrower Holzhauser Lindauer Hansastr Malchower Wuhletalstr HAVEL Heinersdorf Kirche Otisstr Allee Wollankstr JUNGFERNHEIDE Residenzstr Pankow Feldtmannstr Rüdickenstr Max Hermann TEGELER SEE Am Wasserturm M5 Scharnweber Masurenstr M2 Pasedagplatz Berliner Allee Franz Neumann Am Steinberg -

Gesamtverkehrsprognose 2025 Für Die Länder Berlin Und Brandenburg
Abschlussbericht Gesamtverkehrsprognose 2025 für die Länder Berlin und Brandenburg - Ergebnisse - Berlin, 23.11.2009 PTV AG GVP 2025 - Ergebnisse Dokument-Informationen Kurztitel GVP 2025 - Ergebnisse Auftraggeber: Land Berlin Senatsverwaltung für Stadtentwicklung Am Köllnischen Park 3 10179 Berlin Land Brandenburg Ministerium für Infrastruktur und Raumordnung Henning-von-Tresckow-Straße 2-8 14467 Potsdam Auftragnehmer: PTV Planung Transport Verkehr AG Stumpfstraße 1 76131 Karlsruhe TCI Röhling Transport Consulting International Heinrich-Hertz Straße 4 79211 Denzlingen Erstellungsdatum: 23.11.2009 PTV AG Nov/09 GVP 2025 - Ergebnisse Inhalt Inhalt Inhalt ....................................................................................................................... I Tabellen................................................................................................................. III Abbildungen .........................................................................................................IV Abkürzungen ......................................................................................................VIII 1 Veranlassung................................................................................................ 1 2 Grundlagen................................................................................................... 2 2.1 Räumliche Gliederung ...................................................................... 2 2.2 Struktur und Abgrenzung................................................................. -

Train Simulator - Right Through Berlin
Train Simulator - Right through Berlin © Historical S-Bahn Berlin e.V. 2020 Page 1 Train Simulator - Right through Berlin Manual "Mitten durch Berlin" - Operation Series 476 Table of contents Table of contents ....................................................................................................... 2 0. authors ................................................................................................................ 3 1. introduction .......................................................................................................... 3 2. system requirements .............................................................................................. 4 3. the driver's cab ..................................................................................................... 5 4. upgrading .......................................................................................................... 10 4.1 Train is cold (full upgrade) .............................................................................. 10 4.2 Transfer of trains from other Tf ......................................................................... 10 4.3 Train is fully upgraded .................................................................................... 10 4.4 Change of driver's cab ................................................................................... 11 5. door control ....................................................................................................... 11 6. driving and braking ........................................................................................... -

ICON S Conference 17 – 19 June 2016 Humboldt University Berlin
Berlin Berlin 2016 ICON S CONFERENCE 17 – 19 JUNE 2016 HUMBOLDT UNIVERSITY BERLIN We extend a warm welcome to all participants in the 2016 Annual Meeting of ICON S, the International Society of Public Law. This will be our largest Annual Meeting since the foundation of the Society in 2013. The panels, roundtables and plenary events address the Conference’s overarching theme of “Borders, Otherness and Public Law” and other topics at the heart of contemporary public law inquiry. We are grateful to our Berlin hosts for their relentless hard work and creativity in putting together such a mega-sized event, and we thank our sponsors for their generous support. Most of all, we thank you, the ICON S members, for your overwhelmingly positive response to the call for papers this year, and for volunteering your time and energy to promote the success of the Society and its annual conference. Together, we have created what we believe is a first-rate, intellectually appealing program featuring scholars, jurists and policy makers from various disciplines and from literally four corners of the world. We hope that you enjoy it thoroughly! Gráinne de Búrca ( New York University ) and Ran Hirschl ( University of Toronto ) Co-Presidents, ICON S, the International Society of Public Law S Conference ICON WElcomE statEMEnts 1 I WELcoME STATEMENTS 1 II ICON S INAUGURAL GOVERNANCE 3 III SCHEDULE 4 IV MAP OF CONFERENCE VENUES 6 V PLENARY EVENTS 7 VI CONCURRING PANELS 15 a OVERVIEW 16 b PANEls SEssion I 26 c PANEls SEssion II 43 D PANEls SEssion III 77 E PANEls SEssion IV 110 F PANEls SEssion V 143 VII ICON S WP CONFERENCE ProcEEDINGS SERIES 179 VIII SERVICE 180 IX PARTICIPANTS 190 ICON S CONFERENCE 17 – 19 JUNE 2016 HUMBOLDT UNIVERSITY BERLIN We extend a warm welcome to all participants in the 2016 Annual Meeting of ICON S, the International Society of Public Law. -

S-Bahn Linie S46 Fahrpläne & Karten
S-Bahn Linie S46 Fahrpläne & Netzkarten Königs Wusterhausen Bhf ◄ ► Westend Im Website-Modus Anzeigen Die S-Bahn Linie S46 (Königs Wusterhausen Bhf ◄ ► Westend) hat 5 Routen (1) Königs Wusterhausen: 24 Stunden (2) Westend: 24 Stunden Verwende Moovit, um die nächste Station der S-Bahn Linie S46 zu ƒnden und, um zu erfahren wann die nächste S-Bahn Linie S46 kommt. Richtung: Königs Wusterhausen S-Bahn Linie S46 Fahrpläne 23 Haltestellen Abfahrzeiten in Richtung Königs Wusterhausen LINIENPLAN ANZEIGEN Montag 24 Stunden Dienstag 24 Stunden Königs Wusterhausen Bhf Storkower Straße 1, Königs Wusterhausen Mittwoch 24 Stunden Wildau Donnerstag 24 Stunden Bahnhofsplatz 3, Wildau Freitag 24 Stunden Zeuthen Samstag 24 Stunden Spitzbubenweg, Zeuthen Sonntag 24 Stunden Eichwalde Grünau Adlergestell/ Wassersportallee 550, Berlin S-Bahn Linie S46 Info Adlershof Richtung: Königs Wusterhausen Adlergestell 254-256, Berlin Stationen: 23 Fahrtdauer: 57 Min Betriebsbahnhof Schöneweide Linien Informationen: Königs Wusterhausen Bhf, Adlergestell 103, Berlin Wildau, Zeuthen, Eichwalde, Grünau, Adlershof, Betriebsbahnhof Schöneweide, S Schöneweide Bhf, S Schöneweide Bhf Baumschulenweg, Köllnische Heide, Neukölln, Hermannstr., Tempelhof, Südkreuz Bhf, Schöneberg, Baumschulenweg Innsbrucker Platz, Bundesplatz, Heidelberger Platz, S Baumschulenweg, Berlin Hohenzollerndamm, Halensee, Westkreuz, Messe Nord/Icc, Westend Köllnische Heide Krebsgang, Berlin Neukölln Saalestraße 76, Berlin Hermannstr. Hermannstraße, Berlin Tempelhof Tempelhofer Damm 106, Berlin Südkreuz -

WELCOME to LICHTENBERG an Introduction to a District of Berlin
WELCOME TO LICHTENBERG An introduction to a district of Berlin WelcomeENGLISH to Lichtenberg 1 IMPRESSUM (LEGAL DETAILS) Joint editors: District council of Berlin-Lichtenberg, department of staff , economics and fi nancial aff airs, The integrati on commissioner Möllendorff straße 6, 10367 Berlin Internet: www.berlin.de/ba-lichtenberg Associati on in support of the foundati on Wirtschaft skreis Hohenschönhausen-Lichtenberg e.V. Franz-Jacob-Straße 2c, 10369 Berlin Internet: www.wkhl-berlin.de Editorial offi ce: Irina Plat, assistant for refugee matt ers, ph.: 030 90296-3596 Layout/ pictures:: Medienbüro Gäding, Marcel Gäding Zur Alten Börse 77, 12681 Berlin www.medienbuero-gaeding.de pictures in the language guide: Elephant & Castle, Steff en Blankenburg | www.elephant-castle.de Picture credits: p. 1/ p. 3/ p. 39: Marcel Gäding | p. 40-51: Steff en Blankenburg This brochure is funded by the district council of Berlin-Lichtenberg, the associati on in support of the foundati on Wirtschaft skreis Hohenschönhausen-Lichtenberg e.V., Syner- gie GmbH, the housing associati ons Lichtenberg eG (WGLi), Solidarität eG and HOWO- GE. Parts of this brochure, parti cularly the pictures, are subject to copyright. Further distributi on or reuse will therefore need to be agreed upon with the authors in any case. Editorial deadline: January 31st, 2016 The informati on was put together carefully, yet we cannot insure every fact is correct as there may be short-term changes. Sponsors: 2 Willkommen in Lichtenberg Dear new residents of Lichtenberg, message Greeting Welcome to Berlin and welcome to our district of Lichtenberg. Lichtenberg is a part of Berlin, which has altogether about 270,000 inhabitants.