Durham E-Theses
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

SAFETY DATA SHEET Revision Date 28.07.2021 According to Regulation (EC) No
Version 7.0 SAFETY DATA SHEET Revision Date 28.07.2021 according to Regulation (EC) No. 1907/2006 Print Date 02.10.2021 GENERIC EU MSDS - NO COUNTRY SPECIFIC DATA - NO OEL DATA SECTION 1: Identification of the substance/mixture and of the company/undertaking 1.1 Product identifiers Product name : Aluminum iodide Product Number : 208493 Brand : Aldrich REACH No. : A registration number is not available for this substance as the substance or its uses are exempted from registration, the annual tonnage does not require a registration or the registration is envisaged for a later registration deadline. CAS-No. : 7784-23-8 1.2 Relevant identified uses of the substance or mixture and uses advised against Identified uses : Laboratory chemicals, Manufacture of substances 1.3 Details of the supplier of the safety data sheet Company : Sigma-Aldrich Inc. 3050 SPRUCE ST ST. LOUIS MO 63103 UNITED STATES Telephone : +1 314 771-5765 Fax : +1 800 325-5052 1.4 Emergency telephone Emergency Phone # : 800-424-9300 CHEMTREC (USA) +1-703- 527-3887 CHEMTREC (International) 24 Hours/day; 7 Days/week SECTION 2: Hazards identification 2.1 Classification of the substance or mixture Classification according to Regulation (EC) No 1272/2008 Skin corrosion (Sub-category 1B), H314 Serious eye damage (Category 1), H318 For the full text of the H-Statements mentioned in this Section, see Section 16. 2.2 Label elements Labelling according Regulation (EC) No 1272/2008 Aldrich- 208493 Page 1 of 8 The life science business of Merck operates as MilliporeSigma in the US and Canada Pictogram Signal word Danger Hazard statement(s) H314 Causes severe skin burns and eye damage. -

Ep 2508506 A1
(19) & (11) EP 2 508 506 A1 (12) EUROPEAN PATENT APPLICATION (43) Date of publication: (51) Int Cl.: 10.10.2012 Bulletin 2012/41 C07C 67/343 (2006.01) C07C 227/08 (2006.01) C07F 5/02 (2006.01) C07C 229/34 (2006.01) (21) Application number: 11161611.6 (22) Date of filing: 08.04.2011 (84) Designated Contracting States: (72) Inventor: The designation of the inventor has not AL AT BE BG CH CY CZ DE DK EE ES FI FR GB yet been filed GR HR HU IE IS IT LI LT LU LV MC MK MT NL NO PL PT RO RS SE SI SK SM TR (74) Representative: Kunic Tesovic, Barbara Designated Extension States: Lek Pharmaceuticals d.d. BA ME Sandoz Development Center Slovenia - Patents Verovskova 57 (71) Applicant: LEK Pharmaceuticals d.d. 1526 Ljubljana (SI) 1526 Ljubljana (SI) (54) Preparation of sitagliptin intermediates (57) The invention relates to the preparation of chiral compounds, in particular to the preparation of chiral compounds which may be used as intermediates for the preparation of anti-diabetic agents, preferably sitagliptin. EP 2 508 506 A1 Printed by Jouve, 75001 PARIS (FR) EP 2 508 506 A1 Description Field of the Intention 5 [0001] The present invention relates to the preparation of chiral compounds, in particular to the preparation of chiral compounds which may be used as intermediates for the preparation of anti-diabetic agents, preferably sitagliptin. Background prior art 10 [0002] Type II diabetes mellitus (T2DM) is a global epidemic. Therefore, the research is oriented in the development of selective inhibitors of the enzyme DPP-IV as a promising new treatment for the type II diabetes. -
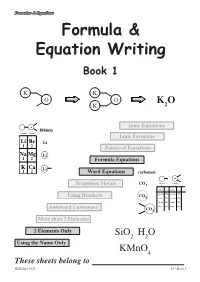
Formula & Equation Writing
Formulae & Equations Formula & Equation Writing Book 1 K K O O K O K 2 Ionic Equations lithium column column 1 2 Ionic Formulae Li Be Li 1 2 Balanced Equations Na Mg Li 1 2 Formula Equations K Ca Li 1 2 Word Equations carbonate valency valency Transition Metals CO3 1 2 name formula name formula ammonium NH4 carbonate CO3 Using Brackets CO 3 cyanide CN chromate CrO4 hydroxide OH sulphate SO4 nitrate NO sulphite SO Awkward Customers 3 3 CO3 More than 2 Elements 2 Elements Only SiO2 H2O Using the Name Only KMnO4 These sheets belong to KHS Sept 2013 page 1 S3 - Book 1 Formulae & Equations What is a Formula ? The formula of a compound tells you two things about the compound :- SiO H O i) which elements are in the compound 2 2 using symbols, and KMnO4 ii) how many atoms of each element are in the compound using numbers. Test Yourself What would be the formula for each of the following? Br H Cl K H C S Al Br O H Cl K H Br Using the Name Only Some compounds have extra information in their carbon monoxide names that allow people to work out and write CO the correct formula. carbon dioxide CO2 The names of the elements appear as usual but dinitrogen tetroxide N O this time the number of each type of atom is 2 4 included using mono- = 1 di- = 12 tri- = 3 tetra- = 4 penta- = 5 hexa- =46 Test Yourself 1 What would be the formula for each of the following? 1. -

Inorganic Syntheses
INORGANIC SYNTHESES Volume 27 .................... ................ Board of Directors JOHN P. FACKLER, JR. Texas A&M University BODlE E. DOUGLAS University of Pittsburgh SMITH L. HOLT, JR. Oklahoma State Uniuersity JAY H. WORRELL University of South Florida RUSSELL N. GRIMES University of Virginia ROBERT J. ANGELIC1 Iowa State University Future Volumes 28 ROBERT J. ANGELIC1 Iowa State University 29 RUSSELL N. GRIMES University of Virginia 30 LEONARD V. INTERRANTE Rensselaer Polytechnic Institute 31 ALLEN H. COWLEY University of Texas, Austin 32 MARCETTA Y. DARENSBOURG Texas A&M University International Associates MARTIN A. BENNETT Australian National University, Canberra FAUSTO CALDERAZZO University of Pisa E. 0. FISCHER Technical University. Munich JACK LEWIS Cambridge University LAMBERTO MALATESTA University of Milan RENE POILBLANC University of Toulouse HERBERT W. ROESKY University of Gottingen F. G. A. STONE University of Bristol GEOFFREY WILKINSON Imperial College of Science and Technology. London AKlO YAMAMOTO Tokyo Institute 01 Technology. Yokohama Editor-in-Chief ALVIN P. GINSBERG INORGANIC SYNTHESES Volume 27 A Wiley-Interscience Publication JOHN WILEY & SONS New York Chichester Brisbane Toronto Singapore A NOTE TO THE READER This book has been electronically reproduced from digital idormation stored at John Wiley h Sons, Inc. We are phased that the use of this new technology will enable us to keep works of enduring scholarly value in print as long as there is a reasonable demand for them. The content of this book is identical to previous printings. Published by John Wiley & Sons, Inc. Copyright $? 1990 Inorganic Syntheses, Inc. All rights reserved. Published simultaneously in Canada. Reproduction or translation of any part of this work beyond that permitted by Section 107 or 108 of the 1976 United States Copyright Act without the permission of the copyright owner is unlawful. -

Pyrophoric Materials
Appendix A PYROPHORIC MATERIALS Pyrophoric materials react with air, or with moisture in air. Typical reactions which occur are oxidation and hydrolysis, and the heat generated by the reactions may ignite the chemical. In some cases, these reactions liberate flammable gases which makes ignition a certainty and explosion a real possibility. Examples of pyrophoric materials are shown below. (List may not be complete) (a) Pyrophoric alkyl metals and derivatives Groups Dodecacarbonyltetracobalt Silver sulphide Dialkytzincs Dodecacarbonyltriiron Sodium disulphide Diplumbanes Hexacarbonylchromium Sodium polysulphide Trialkylaluminiums Hexacarbonylmolybdenum Sodium sulphide Trialkylbismuths Hexacarbonyltungsten Tin (II) sulphide Nonacarbonyldiiron Tin (IV) sulphide Compounds Octacarbonyldicobalt Titanium (IV) sulphide Bis-dimethylstibinyl oxide Pentacarbonyliron Uranium (IV) sulphide Bis(dimethylthallium) acetylide Tetracarbonylnickel Butyllithium (e) Pyrophoric alkyl non-metals Diethylberyllium (c) Pyrophoric metals (finely divided state) Bis-(dibutylborino) acetylene Bis-dimethylarsinyl oxide Diethylcadmium Caesium Rubidium Bis-dimethylarsinyl sulphide Diethylmagnesium Calcium Sodium Bis-trimethylsilyl oxide Diethylzinc Cerium Tantalum Dibutyl-3-methyl-3-buten-1-Yniborane Diisopropylberyllium Chromium Thorium Diethoxydimethylsilane Dimethylberyllium Cobalt Titanium Diethylmethylphosphine Dimethylbismuth chloride Hafnium Uranium Ethyldimthylphosphine Dimethylcadmium Iridium Zirconium Tetraethyldiarsine Dimethylmagnesium Iron Tetramethyldiarsine -

Journal-2019-06
INTELLECTUAL PROPERTY REGISTRY OF SAMOA (IPROS) JOURNAL 2019/06 Publication Date – 4 December 2019 GENERAL INFORMATION The Public is hereby notified that the following persons have applied for the Registration of the Trade Marks as set out in Section 50 of the Intellectual Property Act 2011. Any person who wishes to oppose the registration of any of the under-mentioned Trade Marks, must give notice to the Registrar of Trade Marks, c/- Ministry of Commerce, Industry and Labour, Apia, Samoa within three (3) months from the date of this publication. The notice must be in writing and must include a statement of the grounds of opposition. Each notice of opposition must be accompanied by a filing fee of WST$300. Any person who has any doubts as to whether he/she may be affected by the registration of the Trade Mark in Samoa should consult his/her Solicitor immediately. For any enquiries regarding Trade Marks, please contact the Registry of Intellectual Property at the following telephone number (+685) 20441 or email [email protected] Faafetai, Registrar of Intellectual Property Accepted Trade Marks – Direct Filing (NEWSPAPER VERSION) Application No. : WS/T/7350 Representation of Mark Filing Date : 12 September, 2018 Priority Date : N/A Proprietor/Address : NEST CONSTRUCTION COMPANY LTD Vaitele Uta, Samoa Class(es) 37 Application No. : WS/T/7361 Representation of Mark Filing Date : 24 September, 2018 Priority Date : N/A Proprietor/Address : NIKE INNOVATE C.V. One Bowerman Drive, Beaverton, Oregon 97005--6453, United States of America Class(es) 18 and 25 Application No. : WS/T/7464 Representation of Mark Filing Date : 29 July, 2019 Priority Date : 04/03/2019 Proprietor/Address : REIGN BEVERAGE COMPANY LLC 1547 N. -

Guidelines for Pyrophoric Materials
Guidelines for Pyrophoric Materials Definition and Hazards Pyrophoric materials are substances that ignite instantly upon exposure to air, moisture in the air, oxygen or water. Other common hazards include corrosivity, teratogenicity, and organic peroxide formation, along with damage to the liver, kidneys, and central nervous system. Examples include metal hydrides, finely divided metal powders, nonmetal hydride and alkyl compounds, white phosphorus, alloys of reactive materials and organometallic compounds, including alkylithiums. Additional pyrophoric materials are listed in Appendix A. Failure to follow proper handling techniques could result in serious injury or death. Controlling the Hazards . If possible, use safer chemical alternatives. A “dry run” of the experiment should be performed using low-hazard materials, such as water or solvent, as appropriate. Limit the amount purchased and the amount stored. Do not accumulate unneeded pyrophoric materials. BEFORE working with pyrophoric materials, read the MSDS sheets. The MSDS must be reviewed before using an unfamiliar chemical and periodically as a reminder. A Standard Operating Procedure (SOP) should be prepared and reviewed for each process involving pyrophoric materials. In lab training should be completed and documented. If possible, use the “buddy system”. Working alone with pyrophorics is strongly discouraged. All glassware used for pyrophorics should be oven-dried and free of moisture. Review the location of the safety shower, eyewash, telephone, and fire extinguisher. Keep an appropriate fire extinguisher or extinguishing material close at hand. Additional controls when handling liquid pyrophoric materials . Secure pyrophoric reagent bottle to stand. Secure the syringe so if the plunger blows out of the body of the syringe the contents will not splash anyone. -

(12) Patent Application Publication (10) Pub. No.: US 2015/0175532 A1 Mudduluru Et Al
US 2015O175532A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2015/0175532 A1 Mudduluru et al. (43) Pub. Date: Jun. 25, 2015 (54) PROCESS FOR PREPARATION OF (30) Foreign Application Priority Data 2,3-DIHYDROXY BENZONITRILE Jul. 19, 2012 (IN) ........................... 2926/CHFA2012 (71) Applicant: Laurus Labs Private Limited, Hyderabad (IN) Publication Classification (72) Inventors: Hari Krishna Mudduluru, Hyderabad (51) Int. Cl (IN); Shankar Madhavaram, we Hyderabad (IN) C07C 253/30 (2006.01) (52) U.S. Cl. (73) Assignee: Laurus Labs Private Limited, CPC .................................... C07C 253/30 (2013.01) Hyderabad (IN) (21) Appl. No.: 14/415,527 (57)57 ABSTRACT 1-1. The present invention relates to one pot process for the prepa (22) PCT Filed: Jul.18, 2013 ration of 2,3-dihydroxybenzonitrile from 2,3-dialkoxyben (86). PCT No.: PCT/N2O13AOOO447 Zoic acid without prior isolation of the intermediates. Further the invention relates to the preparation of 2,3-dihydroxyben S371 (c)(1), Zonitrile by dealkylation of 2,3-dialkoxybenzonitrile using (2) Date: Jan. 16, 2015 Suitable aluminum salt-amine complex. US 2015/0175532 A1 Jun. 25, 2015 PROCESS FOR PREPARATION OF 0006. Several reviews have been described for deprotec 2,3-DIHYDROXY BENZONITRILE tion of phenolic ethers. For example, phenolic methyl ethers have deprotected to remove the methyl moiety using hydro PRIORITY gen halide Such as hydrogen chloride or hydrogen bromide under highly acidic conditions; highly dark colored products 0001. This application claims the benefit under Indian Provisional Application No. 2926/CHF/2012 filed 19 Jul. formed in these reactions. In addition the phenolic com 2012 and entitled AN IMPROVED PROCESS FOR THE pounds react further with the halogen compounds used thus PREPARATION OF 2,3-DIHYDROXY BENZONITRILE”, setting a major drawback on this route. -

Chemical Formula of Binary Ionic Compounds – Sheet 1 the Combining Power Or Valency of Silver Is Always 1
Chemical Formula of Binary Ionic Compounds – Sheet 1 The combining power or valency of silver is always 1. All other transition metals are 2 unless otherwise indicated. No. Binary compound Formula No. Binary compound Formula 1 potassium fluoride 26 calcium sulfide 2 calcium chloride 27 lithium bromide 3 barium bromide 28 nickel sulfide 4 silver sulfide 29 zinc phosphide 5 aluminium iodide 30 barium iodide 6 potassium iodide 31 caesium chloride 7 lead(IV) oxide 32 copper bromide 8 zinc nitride 33 sodium nitride 9 silver iodide 34 silver chloride 10 barium fluoride 35 sodium hydride 11 lead(II) iodide 36 potassium nitride 12 silver fluoride 37 cobalt chloride 13 sodium sulfide 38 magnesium sulfide 14 sodium bromide 39 potassium chloride 15 calcium oxide 40 calcium bromide 16 zinc fluoride 41 iron(III) oxide 17 strontium phosphide 42 aluminium fluoride 18 barium sulfide 43 magnesium bromide 19 aluminium oxide 44 iron(III) chloride 20 aluminium chloride 45 barium nitride 21 aluminium sulfide 46 sodium fluoride 22 lead(II) oxide 47 lithium fluoride 23 barium chloride 48 lithium iodide 24 copper chloride 49 lithium hydride 25 barium phosphide 50 potassium oxide “Aluminum” and “cesium” are commonly used alternative spellings for "aluminium" and "caesium that are used in the US. May be freely copied for educational use. ©www.chemicalformula.org Chemical Formula of Binary Ionic Compounds – Sheet 2 The combining power or valency of silver is always 1. All other transition metals are 2 unless otherwise indicated. No. Binary compound Formula No. -

Ep 1831144 B1
(19) & (11) EP 1 831 144 B1 (12) EUROPEAN PATENT SPECIFICATION (45) Date of publication and mention (51) Int Cl.: of the grant of the patent: C07C 37/50 (2006.01) C07C 39/17 (2006.01) 17.08.2011 Bulletin 2011/33 C07C 39/26 (2006.01) (21) Application number: 05816593.7 (86) International application number: PCT/HU2005/000128 (22) Date of filing: 07.12.2005 (87) International publication number: WO 2006/061666 (15.06.2006 Gazette 2006/24) (54) A NEW PROCESS FOR THE PREPARATION OF PHENOLIC HYDROXY-SUBSTITUTED COMPOUNDS NEUES VERFAHREN ZUR HERSTELLUNG PHENOLISCHER HYDROXY SUBSTITUIERTER VERBINDUNGEN PROCEDE D’ELABORATION DE COMPOSES PHENOLIQUES A SUBSTITUTION HYDROXY (84) Designated Contracting States: • BORBÉLY, László AT BE BG CH CY CZ DE DK EE ES FI FR GB GR H-2510 Dorog (HU) HU IE IS IT LI LT LU LV MC NL PL PT RO SE SI • HAÁSZ, Ferenc SK TR H-2500 Esztergom (HU) Designated Extension States: • JEKKEL, Péter AL BA HR MK YU H-2509 Esztergom-Kertváros (HU) (30) Priority: 08.12.2004 HU 0402530 (74) Representative: HOFFMANN EITLE 11.11.2005 HU 0501044 Patent- und Rechtsanwälte Arabellastraße 4 (43) Date of publication of application: 81925 München (DE) 12.09.2007 Bulletin 2007/37 (56) References cited: (73) Proprietor: Richter Gedeon Nyrt. US-A- 4 339 614 1103 Budapest (HU) • MANABU NODE ET.AL.: "Hard acid and soft (72) Inventors: nucleophile system. 2. Demethylation of methyl • VASS, András ethers of alcohol and phenol with an aluminium H-8200 Veszprém (HU) halide-thiolsystem" J. ORG. CHEM., vol. 45, 1980, • DUDÁS, József pages 4275-4277, XP002379759 cited in the H-8200 Veszprém (HU) application Note: Within nine months of the publication of the mention of the grant of the European patent in the European Patent Bulletin, any person may give notice to the European Patent Office of opposition to that patent, in accordance with the Implementing Regulations. -

Chemical Resistance Table
CHEMICAL RESISTANCE TABLE Things to consider when choosing a hose • This chemical resistance table indicates if the inner tube of the hose is resistant to specific materials/chemicals throughout different temperatures. • Some materials/chemicals can change color in contact with the hose. If it´s important to have the color unaffected, we recommend you to contact Hydroscand. • For food products the table only indicates whether the inner tube is resistant to the product. This doesn’t automatically significate that the inner tube is approved for food products. • Abrasion, friction and mechanical influence can increase the chemicals aggression and therefore decrease the durability of the hose. • All values in the table are exclusively for the transport of media. • NB! Materials can change color in contact with other kind of materials. • Outer stress is always an important factor to the durability of the hose. NB! This resistance table should be considered as an indication and not a guarantee. International material codes Rubber Plasic NR Natural rubber P.T.F.E. Polytetraflouro ethylene (Teflon®) SBR Styrene butadiene rubber PP Polypropylene NBR Nitrile rubber UPE Ultra high molekular weight EPDM Ethylene-propylene rubber polyethylene IIR Butyl rubber XLPE Cross linked polyethylene (PEX) CR Chloroprene rubber (Neopren) PU Polyurethane CSM Chlorosulfonated polyethylene PE Polyester platsic (Elastomer) rubber (Hypalon) PA Polyamide (Nylon) PVC Polyvinylchloride How to read the table Fitness grade A Good to excelent B Acceptable for limited use -

List of Reactive Chemicals
LIST OF REACTIVE CHEMICALS Chemical Prefix Chemical Name Reactive Reactive Reactive CAS# Chemical Chemical Chemical Stimulus 1 Stimulus 2 Stimulus 3 111-90-0 "CARBITOL" SOLVENT D 111-15-9 "CELLOSOLVE" ACETATE D 110-80-5 "CELLOSOLVE" SOLVENT D 2- (2,4,6-TRINITROPHENYL)ETHYL ACETATE (1% IN ACETONE & BENZENE S 12427-38-2 AAMANGAN W 88-85-7 AATOX S 40487-42-1 AC 92553 S 105-57-7 ACETAL D 75-07-0 ACETALDEHYDE D 105-57-7 ACETALDEHYDE, DIETHYL ACETAL D 108-05-4 ACETIC ACID ETHENYL ESTER D 108-05-4 ACETIC ACID VINYL ESTER D 75-07-0 ACETIC ALDEHYDE D 101-25-7 ACETO DNPT T 126-84-1 ACETONE DIETHYL ACETAL D 108-05-4 ACETOXYETHYLENE D 108-05-4 1- ACETOXYETHYLENE D 37187-22-7 ACETYL ACETONE PEROXIDE, <=32% AS A PASTE T 37187-22-7 ACETYL ACETONE PEROXIDE, <=42% T 37187-22-7 ACETYL ACETONE PEROXIDE, >42% T S 644-31-5 ACETYL BENZOYL PEROXIDE (SOLID OR MORE THAN 45% IN SOLUTION) T S 644-31-5 ACETYL BENZOYL PEROXIDE, <=45% T 506-96-7 ACETYL BROMIDE W 75-36-5 ACETYL CHLORIDE W ACETYL CYCLOHEXANE SULFONYL PEROXIDE (>82% WITH <12% WATER) T S 3179-56-4 ACETYL CYCLOHEXANE SULFONYL PEROXIDE, <=32% T 3179-56-4 ACETYL CYCLOHEXANE SULFONYL PEROXIDE, <=82% T 674-82-8 ACETYL KETENE (POISON INHALATION HAZARD) D 110-22-5 ACETYL PEROXIDE, <=27% T 110-22-5 ACETYL PEROXIDE, SOLID, OR MORE THAN 27% IN SOLUTION T S 927-86-6 ACETYLCHOLINE PERCHLORATE O S 74-86-2 ACETYLENE D 74-86-2 ACETYLENE (LIQUID) D ACETYLENE SILVER NITRATE D 107-02-08 ACRALDEHYDE (POISON INHALATION HAZARD) D 79-10-7 ACROLEIC ACID D 107-02-08 ACROLEIN, INHIBITED (POISON INHALATION HAZARD) D 107-02-08 ACRYLALDEHYDE (POISON INHALATION HAZARD) D 79-10-7 ACRYLIC ACID D 141-32-2 ACRYLIC ACID BUTYL ESTER D 140-88-5 ACRYLIC ACID ETHYL ESTER D 96-33-3 ACRYLIC ACID METHYL ESTER D Stimulus - Stimuli is the thermal, physical or chemical input needed to induce a hazardous reaction.