Prevotella Multisaccharivorax Type Strain (PPPA20T)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Ninety-Nine De Novo Assembled Genomes from the Moose (Alces Alces) Rumen Microbiome Provide New Insights Into Microbial Plant Biomass Degradation
The ISME Journal (2017) 11, 2538–2551 © 2017 International Society for Microbial Ecology All rights reserved 1751-7362/17 www.nature.com/ismej ORIGINAL ARTICLE Ninety-nine de novo assembled genomes from the moose (Alces alces) rumen microbiome provide new insights into microbial plant biomass degradation Olov Svartström1, Johannes Alneberg2, Nicolas Terrapon3,4, Vincent Lombard3,4, Ino de Bruijn2, Jonas Malmsten5,6, Ann-Marie Dalin6, Emilie EL Muller7, Pranjul Shah7, Paul Wilmes7, Bernard Henrissat3,4,8, Henrik Aspeborg1 and Anders F Andersson2 1School of Biotechnology, Division of Industrial Biotechnology, KTH Royal Institute of Technology, Stockholm, Sweden; 2School of Biotechnology, Division of Gene Technology, KTH Royal Institute of Technology, Science for Life Laboratory, Stockholm, Sweden; 3CNRS UMR 7257, Aix-Marseille University, 13288 Marseille, France; 4INRA, USC 1408 AFMB, 13288 Marseille, France; 5Department of Pathology and Wildlife Diseases, National Veterinary Institute, Uppsala, Sweden; 6Division of Reproduction, Department of Clinical Sciences, Swedish University of Agricultural Sciences, Uppsala, Sweden; 7Luxembourg Centre for Systems Biomedicine, University of Luxembourg, Esch-sur-Alzette, Luxembourg and 8Department of Biological Sciences, King Abdulaziz University, Jeddah, Saudi Arabia The moose (Alces alces) is a ruminant that harvests energy from fiber-rich lignocellulose material through carbohydrate-active enzymes (CAZymes) produced by its rumen microbes. We applied shotgun metagenomics to rumen contents from six moose to obtain insights into this microbiome. Following binning, 99 metagenome-assembled genomes (MAGs) belonging to 11 prokaryotic phyla were reconstructed and characterized based on phylogeny and CAZyme profile. The taxonomy of these MAGs reflected the overall composition of the metagenome, with dominance of the phyla Bacteroidetes and Firmicutes. -

Xiexin Tang Improves the Symptom of Type 2 Diabetic Rats by Modulation of the Gut Microbiota
www.nature.com/scientificreports OPEN Xiexin Tang improves the symptom of type 2 diabetic rats by modulation of the gut microbiota Received: 30 August 2017 Xiaoyan Wei, Jinhua Tao , Suwei Xiao, Shu Jiang, Erxin Shang, Zhenhua Zhu, Dawei Qian Accepted: 13 February 2018 & Jinao Duan Published: xx xx xxxx Type 2 diabetes mellitus (T2DM), a chronic metabolic disease which severely impairs peoples’ quality of life, currently attracted worldwide concerns. There are growing evidences that gut microbiota can exert a great impact on the development of T2DM. Xiexin Tang (XXT), a traditional Chinese medicine prescription, has been clinically used to treat diabetes for thousands of years. However, few researches are investigated on the modulation of gut microbiota community by XXT which will be very helpful to unravel how it works. In this study, bacterial communities were analyzed based on high-throughput 16S rRNA gene sequencing. Results indicated that XXT could notably shape the gut microbiota. T2DM rats treated with XXT exhibited obvious changes in the composition of the gut microbiota, especially for some short chain fatty acids producing and anti-infammatory bacteria such as Adlercreutzia, Alloprevotella, Barnesiella, [Eubacterium] Ventriosum group, Blautia, Lachnospiraceae UCG-001, Papillibacter and Prevotellaceae NK3B31 group. Additionally, XXT could also signifcantly ameliorate hyperglycemia, lipid metabolism dysfunction and infammation in T2DM rats. Moreover, the correlation analysis illustrated that the key microbiota had a close relationship with the T2DM related indexes. The results probably provided useful information for further investigation on its active mechanism and clinical application. T2DM, a chronic metabolic disease characterized by hyperglycemia as a result of insufcient insulin secretion, insulin action or both1, is estimated that its numbers in the adults will increase by 55% by 20352. -

1587714682 277 2.Pdf
Systematic and Applied Microbiology 42 (2019) 107–116 Contents lists available at ScienceDirect Systematic and Applied Microbiology jou rnal homepage: http://www.elsevier.com/locate/syapm The diverse and extensive plant polysaccharide degradative apparatuses of the rumen and hindgut Prevotella species: A factor in their ubiquity? ∗ Tomazˇ Accetto , Gorazd Avgustinˇ University of Ljubljana, Biotechnical faculty, Animal Science Department, Groblje 3, 1230 Domzale,ˇ Slovenia a r t i c l e i n f o a b s t r a c t Article history: Although the Prevotella are commonly observed in high shares in the mammalian hindgut and rumen Received 2 August 2018 studies using NGS approach, the knowledge on their actual role, though postulated to lie in soluble fibre Received in revised form 2 October 2018 degradation, is scarce. Here we analyse in total 23, more than threefold of hitherto known rumen and Accepted 3 October 2018 hindgut Prevotella species and show that rumen/hindgut Prevotella generally possess extensive reper- toires of polysaccharide utilization loci (PULs) and carbohydrate active enzymes targeting various plant Keywords: polysaccharides. These PUL repertoires separate analysed Prevotella into generalists and specialists yet a Prevotella finer diversity among generalists is evident too, both in range of substrates targeted and in PUL combi- Rumen Hindgut nations targeting the same broad substrate classes. Upon evaluation of the shares of species analysed in this study in rumen metagenomes we found firstly, that they contributed significantly to total Prevotella Polysaccharide utilization locus CAZYme abundance though much of rumen Prevotella diversity may still be unknown. Secondly, the hindgut Pre- Metagenome votella species originally isolated in pigs and humans occasionally dominated among the Prevotella with surprisingly high metagenome read shares and were consistently found in rumen metagenome samples from sites as apart as New Zealand and Scotland. -

Gut Microbiome Composition Remains Stable in Individuals with Diabetes-Related Early to Late Stage Chronic Kidney Disease
biomedicines Article Gut Microbiome Composition Remains Stable in Individuals with Diabetes-Related Early to Late Stage Chronic Kidney Disease Ashani Lecamwasam 1,2,3,*, Tiffanie M. Nelson 4, Leni Rivera 3, Elif I. Ekinci 2,5, Richard Saffery 1,6 and Karen M. Dwyer 3 1 Epigenetics Research, Murdoch Children’s Research Institute, VIC 3052, Australia; [email protected] 2 Department of Endocrinology, Austin Health, VIC 3079, Australia; [email protected] 3 School of Medicine, Faculty of Health, Deakin University, VIC 3220, Australia; [email protected] (L.R.); [email protected] (K.M.D.) 4 Menzies Health Institute Queensland, Griffith University, QLD 4222, Australia; [email protected] 5 Department of Medicine, University of Melbourne, VIC 3010, Australia 6 Department of Paediatrics, University of Melbourne, VIC 3010, Australia * Correspondence: [email protected]; Tel.: +613-8341-6200; Fax: +613-9348-1391 Abstract: (1) Background: Individuals with diabetes and chronic kidney disease display gut dysbiosis when compared to healthy controls. However, it is unknown whether there is a change in dysbiosis across the stages of diabetic chronic kidney disease. We investigated a cross-sectional study of patients with early and late diabetes associated chronic kidney disease to identify possible microbial differences between these two groups and across each of the stages of diabetic chronic kidney disease. (2) Methods: This cross-sectional study recruited 95 adults. DNA extracted from collected stool samples were used for 16S rRNA sequencing to identify the bacterial community in the Citation: Lecamwasam, A.; Nelson, gut. (3) Results: The phylum Firmicutes was the most abundant and its mean relative abundance T.M.; Rivera, L.; Ekinci, E.I.; Saffery, was similar in the early and late chronic kidney disease group, 45.99 ± 0.58% and 49.39 ± 0.55%, R.; Dwyer, K.M. -
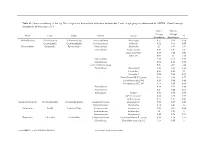
Genus Contributing to the Top 70% of Significant Dissimilarity of Bacteria Between Day 7 and 18 Age Groups As Determined by SIMPER
Table S1: Genus contributing to the top 70% of significant dissimilarity of bacteria between day 7 and 18 age groups as determined by SIMPER. Overall average dissimilarity between ages is 51%. Day 7 Day 18 Average Average Phyla Class Order Family Genus % Abundance Abundance Actinobacteria Actinobacteria Actinomycetales Actinomycetaceae Actinomyces 0.57 0.24 0.74 Coriobacteriia Coriobacteriales Coriobacteriaceae Collinsella 0.51 0.61 0.57 Bacteroidetes Bacteroidia Bacteroidales Bacteroidaceae Bacteroides 2.1 1.63 1.03 Marinifilaceae Butyricimonas 0.82 0.81 0.9 Sanguibacteroides 0.08 0.42 0.59 CAG-873 0.01 1.1 1.68 Marinifilaceae 0.38 0.78 0.91 Marinifilaceae 0.78 1.04 0.97 p-2534-18B5 gut group 0.06 0.7 1.04 Prevotellaceae Alloprevotella 0.46 0.83 0.89 Prevotella 2 0.95 1.11 1.3 Prevotella 7 0.22 0.42 0.56 Prevotellaceae NK3B31 group 0.62 0.68 0.77 Prevotellaceae UCG-003 0.26 0.64 0.89 Prevotellaceae UCG-004 0.11 0.48 0.68 Prevotellaceae 0.64 0.52 0.89 Prevotellaceae 0.27 0.42 0.55 Rikenellaceae Alistipes 0.39 0.62 0.77 dgA-11 gut group 0.04 0.38 0.56 RC9 gut group 0.79 1.17 0.95 Epsilonbacteraeota Campylobacteria Campylobacterales Campylobacteraceae Campylobacter 0.58 0.83 0.97 Helicobacteraceae Helicobacter 0.12 0.41 0.6 Firmicutes Bacilli Lactobacillales Enterococcaceae Enterococcus 0.36 0.31 0.64 Lactobacillaceae Lactobacillus 1.4 1.24 1 Streptococcaceae Streptococcus 0.82 0.58 0.53 Firmicutes Clostridia Clostridiales Christensenellaceae Christensenellaceae R-7 group 0.38 1.36 1.56 Clostridiaceae Clostridium sensu stricto 1 1.16 0.58 -

Role of Actinobacteria and Coriobacteriia in the Antidepressant Effects of Ketamine in an Inflammation Model of Depression
Pharmacology, Biochemistry and Behavior 176 (2019) 93–100 Contents lists available at ScienceDirect Pharmacology, Biochemistry and Behavior journal homepage: www.elsevier.com/locate/pharmbiochembeh Role of Actinobacteria and Coriobacteriia in the antidepressant effects of ketamine in an inflammation model of depression T Niannian Huanga,1, Dongyu Huaa,1, Gaofeng Zhana, Shan Lia, Bin Zhub, Riyue Jiangb, Ling Yangb, ⁎ ⁎ Jiangjiang Bia, Hui Xua, Kenji Hashimotoc, Ailin Luoa, , Chun Yanga, a Department of Anesthesiology, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430030, China b Department of Internal Medicine, The Third Affiliated Hospital of Soochow University, Changzhou 213003, China c Division of Clinical Neuroscience, Chiba University Center for Forensic Mental Health, Chiba 260-8670, Japan ARTICLE INFO ABSTRACT Keywords: Ketamine, an N-methyl-D-aspartic acid receptor (NMDAR) antagonist, elicits rapid-acting and sustained anti- Ketamine depressant effects in treatment-resistant depressed patients. Accumulating evidence suggests that gut microbiota Depression via the gut-brain axis play a role in the pathogenesis of depression, thereby contributing to the antidepressant Lipopolysaccharide actions of certain compounds. Here we investigated the role of gut microbiota in the antidepressant effects of Gut microbiota ketamine in lipopolysaccharide (LPS)-induced inflammation model of depression. Ketamine (10 mg/kg) sig- nificantly attenuated the increased immobility time in forced swimming test (FST), which was associated with the improvements in α-diversity, consisting of Shannon, Simpson and Chao 1 indices. In addition to α-diversity, β-diversity, such as principal coordinates analysis (PCoA), and linear discriminant analysis (LDA) coupled with effect size measurements (LEfSe), showed a differential profile after ketamine treatment. -

Gut Microbiota Differs in Composition and Functionality Between Children
Diabetes Care Volume 41, November 2018 2385 Gut Microbiota Differs in Isabel Leiva-Gea,1 Lidia Sanchez-Alcoholado,´ 2 Composition and Functionality Beatriz Mart´ın-Tejedor,1 Daniel Castellano-Castillo,2,3 Between Children With Type 1 Isabel Moreno-Indias,2,3 Antonio Urda-Cardona,1 Diabetes and MODY2 and Healthy Francisco J. Tinahones,2,3 Jose´ Carlos Fernandez-Garc´ ´ıa,2,3 and Control Subjects: A Case-Control Mar´ıa Isabel Queipo-Ortuno~ 2,3 Study Diabetes Care 2018;41:2385–2395 | https://doi.org/10.2337/dc18-0253 OBJECTIVE Type 1 diabetes is associated with compositional differences in gut microbiota. To date, no microbiome studies have been performed in maturity-onset diabetes of the young 2 (MODY2), a monogenic cause of diabetes. Gut microbiota of type 1 diabetes, MODY2, and healthy control subjects was compared. PATHOPHYSIOLOGY/COMPLICATIONS RESEARCH DESIGN AND METHODS This was a case-control study in 15 children with type 1 diabetes, 15 children with MODY2, and 13 healthy children. Metabolic control and potential factors mod- ifying gut microbiota were controlled. Microbiome composition was determined by 16S rRNA pyrosequencing. 1Pediatric Endocrinology, Hospital Materno- Infantil, Malaga,´ Spain RESULTS 2Clinical Management Unit of Endocrinology and Compared with healthy control subjects, type 1 diabetes was associated with a Nutrition, Laboratory of the Biomedical Research significantly lower microbiota diversity, a significantly higher relative abundance of Institute of Malaga,´ Virgen de la Victoria Uni- Bacteroides Ruminococcus Veillonella Blautia Streptococcus versityHospital,Universidad de Malaga,M´ alaga,´ , , , , and genera, and a Spain lower relative abundance of Bifidobacterium, Roseburia, Faecalibacterium, and 3Centro de Investigacion´ BiomedicaenRed(CIBER)´ Lachnospira. -

Modulation of Gut Microbiota by Glucosamine and Chondroitin in a Randomized, Double-Blind Pilot Trial in Humans
microorganisms Article Modulation of Gut Microbiota by Glucosamine and Chondroitin in a Randomized, Double-Blind Pilot Trial in Humans Sandi L. Navarro 1,* , Lisa Levy 1, Keith R. Curtis 1, Johanna W. Lampe 1,2 and Meredith A.J. Hullar 1 1 Division of Public Health Sciences, Fred Hutchinson Cancer Research Center, Seattle, WA 98109, USA; [email protected] (L.L.); [email protected] (K.R.C.); [email protected] (J.W.L.); [email protected] (M.A.J.H.) 2 Department of Epidemiology, University of Washington, Seattle, WA 98195, USA * Correspondence: [email protected]; Tel.: +1-206-667-6583 Received: 31 October 2019; Accepted: 22 November 2019; Published: 23 November 2019 Abstract: Glucosamine and chondroitin (G&C), typically taken for joint pain, are among the most frequently used specialty supplements by US adults. More recently, G&C have been associated with lower incidence of colorectal cancer in human observational studies and reduced severity of experimentally-induced ulcerative colitis in rodents. However, little is known about their effects on colon-related physiology. G&C are poorly absorbed and therefore metabolized by gut microbiota. G&C have been associated with changes in microbial structure, which may alter host response. We conducted a randomized, double-blind, placebo-controlled crossover trial in ten healthy adults to evaluate the effects of a common dose of G&C compared to placebo for 14 days on gut microbial community structure, measured by 16S rRNA gene sequencing. Linear mixed models were used to evaluate the effect of G&C compared to placebo on fecal microbial alpha and beta diversity, seven phyla, and 137 genera. -

Short-Chain Fatty Acids Modulate Metabolic Pathways and Membrane Lipids in Prevotella Bryantii B14
proteomes Article Short-Chain Fatty Acids Modulate Metabolic Pathways and Membrane Lipids in Prevotella bryantii B14 Andrej Trautmann 1, Lena Schleicher 2, Simon Deusch 1 , Jochem Gätgens 3 , Julia Steuber 2 and Jana Seifert 1,* 1 Institute of Animal Science, University of Hohenheim, 70599 Stuttgart, Germany; [email protected] (A.T.); [email protected] (S.D.) 2 Institute of Biology, University of Hohenheim, 70599 Stuttgart, Germany; [email protected] (L.S.); [email protected] (J.S.) 3 Institute of Bio- and Geosciences, IBG-1: Biotechnology, Forschungszentrum Jülich, 52425 Jülich, Germany; [email protected] * Correspondence: [email protected] Received: 22 September 2020; Accepted: 13 October 2020; Published: 16 October 2020 Abstract: Short-chain fatty acids (SCFAs) are bacterial products that are known to be used as energy sources in eukaryotic hosts, whereas their role in the metabolism of intestinal microbes is rarely explored. In the present study, acetic, propionic, butyric, isobutyric, valeric, and isovaleric acid, respectively, were added to a newly defined medium containing Prevotella bryantii B14 cells. After 8 h and 24 h, optical density, pH and SCFA concentrations were measured. Long-chain fatty acid (LCFA) profiles of the bacterial cells were analyzed via gas chromatography-time of flight-mass spectrometry (GC-ToF MS) and proteins were quantified using a mass spectrometry-based, label-free approach. Cultures supplemented with single SCFAs revealed different growth behavior. Structural features of the respective SCFAs were identified in the LCFA profiles, which suggests incorporation into the bacterial membranes. The proteomes of cultures supplemented with acetic and valeric acid differed by an increased abundance of outer membrane proteins. -

Characterization of Antibiotic Resistance Genes in the Species of the Rumen Microbiota
ARTICLE https://doi.org/10.1038/s41467-019-13118-0 OPEN Characterization of antibiotic resistance genes in the species of the rumen microbiota Yasmin Neves Vieira Sabino1, Mateus Ferreira Santana1, Linda Boniface Oyama2, Fernanda Godoy Santos2, Ana Júlia Silva Moreira1, Sharon Ann Huws2* & Hilário Cuquetto Mantovani 1* Infections caused by multidrug resistant bacteria represent a therapeutic challenge both in clinical settings and in livestock production, but the prevalence of antibiotic resistance genes 1234567890():,; among the species of bacteria that colonize the gastrointestinal tract of ruminants is not well characterized. Here, we investigate the resistome of 435 ruminal microbial genomes in silico and confirm representative phenotypes in vitro. We find a high abundance of genes encoding tetracycline resistance and evidence that the tet(W) gene is under positive selective pres- sure. Our findings reveal that tet(W) is located in a novel integrative and conjugative element in several ruminal bacterial genomes. Analyses of rumen microbial metatranscriptomes confirm the expression of the most abundant antibiotic resistance genes. Our data provide insight into antibiotic resistange gene profiles of the main species of ruminal bacteria and reveal the potential role of mobile genetic elements in shaping the resistome of the rumen microbiome, with implications for human and animal health. 1 Departamento de Microbiologia, Universidade Federal de Viçosa, Viçosa, Minas Gerais, Brazil. 2 Institute for Global Food Security, School of Biological -

Product Sheet Info
Product Information Sheet for HM-271 Prevotella nigrescens, Strain F0103 Tryptic Soy Agar with 5% sheep blood or equivalent Incubation: Catalog No. HM-271 Temperature: 37°C Atmosphere: Anaerobic (80% N :10% CO :10% H ) 2 2 2 Propagation: For research use only. Not for human use. 1. Keep vial frozen until ready for use, then thaw. 2. Transfer the entire thawed aliquot into a single tube of Contributor: broth. Jacques Izard, Assistant Member of the Staff, Department of 3. Use several drops of the suspension to inoculate an Molecular Genetics, The Forsyth Institute, Boston, agar slant and/or plate. Massachusetts 4. Incubate the tube, slant and/or plate at 37°C for 48 to 72 hours. Manufacturer: BEI Resources Citation: Acknowledgment for publications should read “The following Product Description: reagent was obtained through BEI Resources, NIAID, NIH as Bacteria Classification: Prevotellaceae, Prevotella part of the Human Microbiome Project: Prevotella Species: Prevotella nigrescens nigrescens, Strain F0103, HM-271.” Strain: F0103 Original Source: Prevotella nigrescens (P. nigrescens), Biosafety Level: 2 1 strain F0103 was isolated from a human oral cavity. Appropriate safety procedures should always be used with Comments: P. nigrescens, strain F0103 (HMP ID 0662) is a this material. Laboratory safety is discussed in the following reference genome for The Human Microbiome Project publication: U.S. Department of Health and Human Services, (HMP). HMP is an initiative to identify and characterize Public Health Service, Centers for Disease Control and human microbial flora. The complete genome of P. Prevention, and National Institutes of Health. Biosafety in nigrescens, strain F0103 is currently being sequenced at Microbiological and Biomedical Laboratories. -

Db20-0503.Full.Pdf
Page 1 of 32 Diabetes Analysis of the Composition and Functions of the Microbiome in Diabetic Foot Osteomyelitis based on 16S rRNA and Metagenome Sequencing Technology Zou Mengchen1*; Cai Yulan2*; Hu Ping3*; Cao Yin1; Luo Xiangrong1; Fan Xinzhao1; Zhang Bao4; Wu Xianbo4; Jiang Nan5; Lin Qingrong5; Zhou Hao6; Xue Yaoming1; Gao Fang1# 1Department of Endocrinology and Metabolism, Nanfang Hospital, Southern Medical University, Guangzhou, China 2Department of Endocrinology, Affiliated Hospital of Zunyi Medical College, Zunyi, China 3Department of Geriatric Medicine, Xiaogan Central Hospital, Xiaogan, China 4School of Public Health and Tropic Medicine, Southern Medical University, Guangzhou, China 5Department of Orthopaedics & Traumatology, Nanfang Hospital, Southern Medical University, Guangzhou, China 6Department of Hospital Infection Management of Nanfang Hospital, Southern Medical University, Guangzhou, China *Zou mengchen, Cai yulan and Hu ping contributed equally to this work. Running title: Microbiome of Diabetic Foot Osteomyelitis Word count: 3915 Figures/Tables Count: 4Figures / 3 Tables References: 26 Diabetes Publish Ahead of Print, published online August 14, 2020 Diabetes Page 2 of 32 Keywords: diabetic foot osteomyelitis; microbiome; 16S rRNA sequencing; metagenome sequencing #Corresponding author: Gao Fang, E-mail: [email protected], Tel: 13006871226 Page 3 of 32 Diabetes ABSTRACT Metagenome sequencing has not been used in infected bone specimens. This study aimed to analyze the microbiome and its functions. This prospective observational study explored the microbiome and its functions of DFO (group DM) and posttraumatic foot osteomyelitis (PFO) (group NDM) based on 16S rRNA sequencing and metagenome sequencing technologies. Spearman analysis was used to explore the correlation between dominant species and clinical indicators of patients with DFO.