Association of Dietary Patterns with the Fecal Microbiota in Korean Adolescents Han Byul Jang1, Min-Kyu Choi2, Jae Heon Kang3, Sang Ick Park1 and Hye-Ja Lee1*
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Xiexin Tang Improves the Symptom of Type 2 Diabetic Rats by Modulation of the Gut Microbiota
www.nature.com/scientificreports OPEN Xiexin Tang improves the symptom of type 2 diabetic rats by modulation of the gut microbiota Received: 30 August 2017 Xiaoyan Wei, Jinhua Tao , Suwei Xiao, Shu Jiang, Erxin Shang, Zhenhua Zhu, Dawei Qian Accepted: 13 February 2018 & Jinao Duan Published: xx xx xxxx Type 2 diabetes mellitus (T2DM), a chronic metabolic disease which severely impairs peoples’ quality of life, currently attracted worldwide concerns. There are growing evidences that gut microbiota can exert a great impact on the development of T2DM. Xiexin Tang (XXT), a traditional Chinese medicine prescription, has been clinically used to treat diabetes for thousands of years. However, few researches are investigated on the modulation of gut microbiota community by XXT which will be very helpful to unravel how it works. In this study, bacterial communities were analyzed based on high-throughput 16S rRNA gene sequencing. Results indicated that XXT could notably shape the gut microbiota. T2DM rats treated with XXT exhibited obvious changes in the composition of the gut microbiota, especially for some short chain fatty acids producing and anti-infammatory bacteria such as Adlercreutzia, Alloprevotella, Barnesiella, [Eubacterium] Ventriosum group, Blautia, Lachnospiraceae UCG-001, Papillibacter and Prevotellaceae NK3B31 group. Additionally, XXT could also signifcantly ameliorate hyperglycemia, lipid metabolism dysfunction and infammation in T2DM rats. Moreover, the correlation analysis illustrated that the key microbiota had a close relationship with the T2DM related indexes. The results probably provided useful information for further investigation on its active mechanism and clinical application. T2DM, a chronic metabolic disease characterized by hyperglycemia as a result of insufcient insulin secretion, insulin action or both1, is estimated that its numbers in the adults will increase by 55% by 20352. -

Gut Microbiome Composition Remains Stable in Individuals with Diabetes-Related Early to Late Stage Chronic Kidney Disease
biomedicines Article Gut Microbiome Composition Remains Stable in Individuals with Diabetes-Related Early to Late Stage Chronic Kidney Disease Ashani Lecamwasam 1,2,3,*, Tiffanie M. Nelson 4, Leni Rivera 3, Elif I. Ekinci 2,5, Richard Saffery 1,6 and Karen M. Dwyer 3 1 Epigenetics Research, Murdoch Children’s Research Institute, VIC 3052, Australia; [email protected] 2 Department of Endocrinology, Austin Health, VIC 3079, Australia; [email protected] 3 School of Medicine, Faculty of Health, Deakin University, VIC 3220, Australia; [email protected] (L.R.); [email protected] (K.M.D.) 4 Menzies Health Institute Queensland, Griffith University, QLD 4222, Australia; [email protected] 5 Department of Medicine, University of Melbourne, VIC 3010, Australia 6 Department of Paediatrics, University of Melbourne, VIC 3010, Australia * Correspondence: [email protected]; Tel.: +613-8341-6200; Fax: +613-9348-1391 Abstract: (1) Background: Individuals with diabetes and chronic kidney disease display gut dysbiosis when compared to healthy controls. However, it is unknown whether there is a change in dysbiosis across the stages of diabetic chronic kidney disease. We investigated a cross-sectional study of patients with early and late diabetes associated chronic kidney disease to identify possible microbial differences between these two groups and across each of the stages of diabetic chronic kidney disease. (2) Methods: This cross-sectional study recruited 95 adults. DNA extracted from collected stool samples were used for 16S rRNA sequencing to identify the bacterial community in the Citation: Lecamwasam, A.; Nelson, gut. (3) Results: The phylum Firmicutes was the most abundant and its mean relative abundance T.M.; Rivera, L.; Ekinci, E.I.; Saffery, was similar in the early and late chronic kidney disease group, 45.99 ± 0.58% and 49.39 ± 0.55%, R.; Dwyer, K.M. -
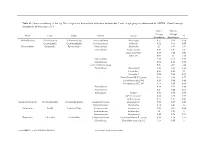
Genus Contributing to the Top 70% of Significant Dissimilarity of Bacteria Between Day 7 and 18 Age Groups As Determined by SIMPER
Table S1: Genus contributing to the top 70% of significant dissimilarity of bacteria between day 7 and 18 age groups as determined by SIMPER. Overall average dissimilarity between ages is 51%. Day 7 Day 18 Average Average Phyla Class Order Family Genus % Abundance Abundance Actinobacteria Actinobacteria Actinomycetales Actinomycetaceae Actinomyces 0.57 0.24 0.74 Coriobacteriia Coriobacteriales Coriobacteriaceae Collinsella 0.51 0.61 0.57 Bacteroidetes Bacteroidia Bacteroidales Bacteroidaceae Bacteroides 2.1 1.63 1.03 Marinifilaceae Butyricimonas 0.82 0.81 0.9 Sanguibacteroides 0.08 0.42 0.59 CAG-873 0.01 1.1 1.68 Marinifilaceae 0.38 0.78 0.91 Marinifilaceae 0.78 1.04 0.97 p-2534-18B5 gut group 0.06 0.7 1.04 Prevotellaceae Alloprevotella 0.46 0.83 0.89 Prevotella 2 0.95 1.11 1.3 Prevotella 7 0.22 0.42 0.56 Prevotellaceae NK3B31 group 0.62 0.68 0.77 Prevotellaceae UCG-003 0.26 0.64 0.89 Prevotellaceae UCG-004 0.11 0.48 0.68 Prevotellaceae 0.64 0.52 0.89 Prevotellaceae 0.27 0.42 0.55 Rikenellaceae Alistipes 0.39 0.62 0.77 dgA-11 gut group 0.04 0.38 0.56 RC9 gut group 0.79 1.17 0.95 Epsilonbacteraeota Campylobacteria Campylobacterales Campylobacteraceae Campylobacter 0.58 0.83 0.97 Helicobacteraceae Helicobacter 0.12 0.41 0.6 Firmicutes Bacilli Lactobacillales Enterococcaceae Enterococcus 0.36 0.31 0.64 Lactobacillaceae Lactobacillus 1.4 1.24 1 Streptococcaceae Streptococcus 0.82 0.58 0.53 Firmicutes Clostridia Clostridiales Christensenellaceae Christensenellaceae R-7 group 0.38 1.36 1.56 Clostridiaceae Clostridium sensu stricto 1 1.16 0.58 -

Role of Actinobacteria and Coriobacteriia in the Antidepressant Effects of Ketamine in an Inflammation Model of Depression
Pharmacology, Biochemistry and Behavior 176 (2019) 93–100 Contents lists available at ScienceDirect Pharmacology, Biochemistry and Behavior journal homepage: www.elsevier.com/locate/pharmbiochembeh Role of Actinobacteria and Coriobacteriia in the antidepressant effects of ketamine in an inflammation model of depression T Niannian Huanga,1, Dongyu Huaa,1, Gaofeng Zhana, Shan Lia, Bin Zhub, Riyue Jiangb, Ling Yangb, ⁎ ⁎ Jiangjiang Bia, Hui Xua, Kenji Hashimotoc, Ailin Luoa, , Chun Yanga, a Department of Anesthesiology, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430030, China b Department of Internal Medicine, The Third Affiliated Hospital of Soochow University, Changzhou 213003, China c Division of Clinical Neuroscience, Chiba University Center for Forensic Mental Health, Chiba 260-8670, Japan ARTICLE INFO ABSTRACT Keywords: Ketamine, an N-methyl-D-aspartic acid receptor (NMDAR) antagonist, elicits rapid-acting and sustained anti- Ketamine depressant effects in treatment-resistant depressed patients. Accumulating evidence suggests that gut microbiota Depression via the gut-brain axis play a role in the pathogenesis of depression, thereby contributing to the antidepressant Lipopolysaccharide actions of certain compounds. Here we investigated the role of gut microbiota in the antidepressant effects of Gut microbiota ketamine in lipopolysaccharide (LPS)-induced inflammation model of depression. Ketamine (10 mg/kg) sig- nificantly attenuated the increased immobility time in forced swimming test (FST), which was associated with the improvements in α-diversity, consisting of Shannon, Simpson and Chao 1 indices. In addition to α-diversity, β-diversity, such as principal coordinates analysis (PCoA), and linear discriminant analysis (LDA) coupled with effect size measurements (LEfSe), showed a differential profile after ketamine treatment. -

The Phylogenetic Composition and Structure of Soil Microbial Communities Shifts in Response to Elevated Carbon Dioxide
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by University of Minnesota Digital Conservancy The ISME Journal (2012) 6, 259–272 & 2012 International Society for Microbial Ecology All rights reserved 1751-7362/12 www.nature.com/ismej ORIGINAL ARTICLE The phylogenetic composition and structure of soil microbial communities shifts in response to elevated carbon dioxide Zhili He1, Yvette Piceno2, Ye Deng1, Meiying Xu1,3, Zhenmei Lu1,4, Todd DeSantis2, Gary Andersen2, Sarah E Hobbie5, Peter B Reich6 and Jizhong Zhou1,2 1Institute for Environmental Genomics, Department of Botany and Microbiology, University of Oklahoma, Norman, OK, USA; 2Ecology Department, Earth Sciences Division, Lawrence Berkeley National Laboratory, Berkeley, CA, USA; 3Guangdong Provincial Key Laboratory of Microbial Culture Collection and Application, Guangdong Institute of Microbiology, Guangzhou, China; 4College of Life Sciences, Zhejiang University, Hangzhou, China; 5Department of Ecology, Evolution, and Behavior, St Paul, MN, USA and 6Department of Forest Resources, University of Minnesota, St Paul, MN, USA One of the major factors associated with global change is the ever-increasing concentration of atmospheric CO2. Although the stimulating effects of elevated CO2 (eCO2) on plant growth and primary productivity have been established, its impacts on the diversity and function of soil microbial communities are poorly understood. In this study, phylogenetic microarrays (PhyloChip) were used to comprehensively survey the richness, composition and structure of soil microbial communities in a grassland experiment subjected to two CO2 conditions (ambient, 368 p.p.m., versus elevated, 560 p.p.m.) for 10 years. The richness based on the detected number of operational taxonomic units (OTUs) significantly decreased under eCO2. -

Gut Microbiota Differs in Composition and Functionality Between Children
Diabetes Care Volume 41, November 2018 2385 Gut Microbiota Differs in Isabel Leiva-Gea,1 Lidia Sanchez-Alcoholado,´ 2 Composition and Functionality Beatriz Mart´ın-Tejedor,1 Daniel Castellano-Castillo,2,3 Between Children With Type 1 Isabel Moreno-Indias,2,3 Antonio Urda-Cardona,1 Diabetes and MODY2 and Healthy Francisco J. Tinahones,2,3 Jose´ Carlos Fernandez-Garc´ ´ıa,2,3 and Control Subjects: A Case-Control Mar´ıa Isabel Queipo-Ortuno~ 2,3 Study Diabetes Care 2018;41:2385–2395 | https://doi.org/10.2337/dc18-0253 OBJECTIVE Type 1 diabetes is associated with compositional differences in gut microbiota. To date, no microbiome studies have been performed in maturity-onset diabetes of the young 2 (MODY2), a monogenic cause of diabetes. Gut microbiota of type 1 diabetes, MODY2, and healthy control subjects was compared. PATHOPHYSIOLOGY/COMPLICATIONS RESEARCH DESIGN AND METHODS This was a case-control study in 15 children with type 1 diabetes, 15 children with MODY2, and 13 healthy children. Metabolic control and potential factors mod- ifying gut microbiota were controlled. Microbiome composition was determined by 16S rRNA pyrosequencing. 1Pediatric Endocrinology, Hospital Materno- Infantil, Malaga,´ Spain RESULTS 2Clinical Management Unit of Endocrinology and Compared with healthy control subjects, type 1 diabetes was associated with a Nutrition, Laboratory of the Biomedical Research significantly lower microbiota diversity, a significantly higher relative abundance of Institute of Malaga,´ Virgen de la Victoria Uni- Bacteroides Ruminococcus Veillonella Blautia Streptococcus versityHospital,Universidad de Malaga,M´ alaga,´ , , , , and genera, and a Spain lower relative abundance of Bifidobacterium, Roseburia, Faecalibacterium, and 3Centro de Investigacion´ BiomedicaenRed(CIBER)´ Lachnospira. -

Modulation of Gut Microbiota by Glucosamine and Chondroitin in a Randomized, Double-Blind Pilot Trial in Humans
microorganisms Article Modulation of Gut Microbiota by Glucosamine and Chondroitin in a Randomized, Double-Blind Pilot Trial in Humans Sandi L. Navarro 1,* , Lisa Levy 1, Keith R. Curtis 1, Johanna W. Lampe 1,2 and Meredith A.J. Hullar 1 1 Division of Public Health Sciences, Fred Hutchinson Cancer Research Center, Seattle, WA 98109, USA; [email protected] (L.L.); [email protected] (K.R.C.); [email protected] (J.W.L.); [email protected] (M.A.J.H.) 2 Department of Epidemiology, University of Washington, Seattle, WA 98195, USA * Correspondence: [email protected]; Tel.: +1-206-667-6583 Received: 31 October 2019; Accepted: 22 November 2019; Published: 23 November 2019 Abstract: Glucosamine and chondroitin (G&C), typically taken for joint pain, are among the most frequently used specialty supplements by US adults. More recently, G&C have been associated with lower incidence of colorectal cancer in human observational studies and reduced severity of experimentally-induced ulcerative colitis in rodents. However, little is known about their effects on colon-related physiology. G&C are poorly absorbed and therefore metabolized by gut microbiota. G&C have been associated with changes in microbial structure, which may alter host response. We conducted a randomized, double-blind, placebo-controlled crossover trial in ten healthy adults to evaluate the effects of a common dose of G&C compared to placebo for 14 days on gut microbial community structure, measured by 16S rRNA gene sequencing. Linear mixed models were used to evaluate the effect of G&C compared to placebo on fecal microbial alpha and beta diversity, seven phyla, and 137 genera. -

Short-Chain Fatty Acids Modulate Metabolic Pathways and Membrane Lipids in Prevotella Bryantii B14
proteomes Article Short-Chain Fatty Acids Modulate Metabolic Pathways and Membrane Lipids in Prevotella bryantii B14 Andrej Trautmann 1, Lena Schleicher 2, Simon Deusch 1 , Jochem Gätgens 3 , Julia Steuber 2 and Jana Seifert 1,* 1 Institute of Animal Science, University of Hohenheim, 70599 Stuttgart, Germany; [email protected] (A.T.); [email protected] (S.D.) 2 Institute of Biology, University of Hohenheim, 70599 Stuttgart, Germany; [email protected] (L.S.); [email protected] (J.S.) 3 Institute of Bio- and Geosciences, IBG-1: Biotechnology, Forschungszentrum Jülich, 52425 Jülich, Germany; [email protected] * Correspondence: [email protected] Received: 22 September 2020; Accepted: 13 October 2020; Published: 16 October 2020 Abstract: Short-chain fatty acids (SCFAs) are bacterial products that are known to be used as energy sources in eukaryotic hosts, whereas their role in the metabolism of intestinal microbes is rarely explored. In the present study, acetic, propionic, butyric, isobutyric, valeric, and isovaleric acid, respectively, were added to a newly defined medium containing Prevotella bryantii B14 cells. After 8 h and 24 h, optical density, pH and SCFA concentrations were measured. Long-chain fatty acid (LCFA) profiles of the bacterial cells were analyzed via gas chromatography-time of flight-mass spectrometry (GC-ToF MS) and proteins were quantified using a mass spectrometry-based, label-free approach. Cultures supplemented with single SCFAs revealed different growth behavior. Structural features of the respective SCFAs were identified in the LCFA profiles, which suggests incorporation into the bacterial membranes. The proteomes of cultures supplemented with acetic and valeric acid differed by an increased abundance of outer membrane proteins. -

Novel Molecular, Structural and Evolutionary Characteristics of the Phosphoketolases from Bifidobacteria and Coriobacteriales
RESEARCH ARTICLE Novel molecular, structural and evolutionary characteristics of the phosphoketolases from bifidobacteria and Coriobacteriales Radhey S. Gupta*, Anish Nanda, Bijendra Khadka Department of Biochemistry and Biomedical Sciences, McMaster University, Hamilton, Ontario, Canada * [email protected] a1111111111 a1111111111 a1111111111 Abstract a1111111111 Members from the order Bifidobacteriales, which include many species exhibiting health a1111111111 promoting effects, differ from all other organisms in using a unique pathway for carbohydrate metabolism, known as the ªbifid shuntº, which utilizes the enzyme phosphoketolase (PK) to carry out the phosphorolysis of both fructose-6-phosphate (F6P) and xylulose-5-phosphate (X5P). In contrast to bifidobacteria, the PKs found in other organisms (referred to XPK) are OPEN ACCESS able to metabolize primarily X5P and show very little activity towards F6P. Presently, very lit- Citation: Gupta RS, Nanda A, Khadka B (2017) tle is known about the molecular or biochemical basis of the differences in the two forms of Novel molecular, structural and evolutionary PKs. Comparative analyses of PK sequences from different organisms reported here have characteristics of the phosphoketolases from bifidobacteria and Coriobacteriales. PLoS ONE 12 identified multiple high-specific sequence features in the forms of conserved signature (2): e0172176. doi:10.1371/journal.pone.0172176 inserts and deletions (CSIs) in the PK sequences that clearly distinguish the X5P/F6P phos- Editor: Eugene A. Permyakov, Russian Academy of phoketolases (XFPK) of bifidobacteria from the XPK homologs found in most other organ- Medical Sciences, RUSSIAN FEDERATION isms. Interestingly, most of the molecular signatures that are specific for the XFPK from Received: December 12, 2016 bifidobacteria are also shared by the PK homologs from the Coriobacteriales order of Acti- nobacteria. -

Bacterial Diversity and Functional Analysis of Severe Early Childhood
www.nature.com/scientificreports OPEN Bacterial diversity and functional analysis of severe early childhood caries and recurrence in India Balakrishnan Kalpana1,3, Puniethaa Prabhu3, Ashaq Hussain Bhat3, Arunsaikiran Senthilkumar3, Raj Pranap Arun1, Sharath Asokan4, Sachin S. Gunthe2 & Rama S. Verma1,5* Dental caries is the most prevalent oral disease afecting nearly 70% of children in India and elsewhere. Micro-ecological niche based acidifcation due to dysbiosis in oral microbiome are crucial for caries onset and progression. Here we report the tooth bacteriome diversity compared in Indian children with caries free (CF), severe early childhood caries (SC) and recurrent caries (RC). High quality V3–V4 amplicon sequencing revealed that SC exhibited high bacterial diversity with unique combination and interrelationship. Gracillibacteria_GN02 and TM7 were unique in CF and SC respectively, while Bacteroidetes, Fusobacteria were signifcantly high in RC. Interestingly, we found Streptococcus oralis subsp. tigurinus clade 071 in all groups with signifcant abundance in SC and RC. Positive correlation between low and high abundant bacteria as well as with TCS, PTS and ABC transporters were seen from co-occurrence network analysis. This could lead to persistence of SC niche resulting in RC. Comparative in vitro assessment of bioflm formation showed that the standard culture of S. oralis and its phylogenetically similar clinical isolates showed profound bioflm formation and augmented the growth and enhanced bioflm formation in S. mutans in both dual and multispecies cultures. Interaction among more than 700 species of microbiota under diferent micro-ecological niches of the human oral cavity1,2 acts as a primary defense against various pathogens. Tis has been observed to play a signifcant role in child’s oral and general health. -

Wedding Higher Taxonomic Ranks with Metabolic Signatures Coded in Prokaryotic Genomes
Wedding higher taxonomic ranks with metabolic signatures coded in prokaryotic genomes Gregorio Iraola*, Hugo Naya* Corresponding authors: E-mail: [email protected], [email protected] This PDF file includes: Supplementary Table 1 Supplementary Figures 1 to 4 Supplementary Methods SUPPLEMENTARY TABLES Supplementary Tab. 1 Supplementary Tab. 1. Full prediction for the set of 108 external genomes used as test. genome domain phylum class order family genus prediction alphaproteobacterium_LFTY0 Bacteria Proteobacteria Alphaproteobacteria Rhodobacterales Rhodobacteraceae Unknown candidatus_nasuia_deltocephalinicola_PUNC_CP013211 Bacteria Proteobacteria Gammaproteobacteria Unknown Unknown Unknown candidatus_sulcia_muelleri_PUNC_CP013212 Bacteria Bacteroidetes Flavobacteriia Flavobacteriales NA Candidatus Sulcia deinococcus_grandis_ATCC43672_BCMS0 Bacteria Deinococcus-Thermus Deinococci Deinococcales Deinococcaceae Deinococcus devosia_sp_H5989_CP011300 Bacteria Proteobacteria Unknown Unknown Unknown Unknown micromonospora_RV43_LEKG0 Bacteria Actinobacteria Actinobacteria Micromonosporales Micromonosporaceae Micromonospora nitrosomonas_communis_Nm2_CP011451 Bacteria Proteobacteria Betaproteobacteria Nitrosomonadales Nitrosomonadaceae Unknown nocardia_seriolae_U1_BBYQ0 Bacteria Actinobacteria Actinobacteria Corynebacteriales Nocardiaceae Nocardia nocardiopsis_RV163_LEKI01 Bacteria Actinobacteria Actinobacteria Streptosporangiales Nocardiopsaceae Nocardiopsis oscillatoriales_cyanobacterium_MTP1_LNAA0 Bacteria Cyanobacteria NA Oscillatoriales -

Prevotella Multisaccharivorax Type Strain (PPPA20T)
Lawrence Berkeley National Laboratory Recent Work Title Non-contiguous finished genome sequence of the opportunistic oral pathogen Prevotella multisaccharivorax type strain (PPPA20). Permalink https://escholarship.org/uc/item/0p79h5ds Journal Standards in genomic sciences, 5(1) ISSN 1944-3277 Authors Pati, Amrita Gronow, Sabine Lu, Megan et al. Publication Date 2011-10-01 DOI 10.4056/sigs.2164949 Peer reviewed eScholarship.org Powered by the California Digital Library University of California Standards in Genomic Sciences (2011) 5:41-49 DOI:10.4056/sigs.2164949 Non-contiguous finished genome sequence of the opportunistic oral pathogen Prevotella multisaccharivorax type strain (PPPA20T) Amrita Pati1, Sabine Gronow2, Megan Lu1,3, Alla Lapidus1, Matt Nolan1, Susan Lucas1, Nancy Hammon1, Shweta Deshpande1, Jan-Fang Cheng1, Roxanne Tapia1,3, Cliff Han1,3, Lynne Goodwin1,3 Sam Pitluck1, Konstantinos Liolios1, Ioanna Pagani1, Konstantinos Mavromatis1, Natalia Mikhailova1, Marcel Huntemann1, Amy Chen4, Krishna Palaniappan4, Miriam Land1,5, Loren Hauser1,5, John C. Detter1,3, Evelyne-Marie Brambilla2, Manfred Rohde6, Markus Göker2, Tanja Woyke1, James Bristow1, Jonathan A. Eisen1,7, Victor Markowitz4, Philip Hugenholtz1,8, Nikos C. Kyrpides1, Hans-Peter Klenk2*, and Natalia Ivanova1 1 DOE Joint Genome Institute, Walnut Creek, California, USA 2 DSMZ - German Collection of Microorganisms and Cell Cultures GmbH, Braunschweig, Germany 3 Los Alamos National Laboratory, Bioscience Division, Los Alamos, New Mexico, USA 4 Biological Data Management and