Copy Number Increases of Transposable Elements and Protein-Coding Genes in an Invasive Fish of Hybrid Origin
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Die Nase (Chondrostoma Nasus) Im Einzugsgebiet Des Bodensees – Grundlagenbericht 1
Die Nase (Chondrostoma nasus) im Einzugsgebiet des Bodensees – Grundlagenbericht 1 Die Nase (Chondrostoma nasus) im Einzugsgebiet des Bodensees Grundlagenbericht für internationale Maßnahmenprogramme HYDRA Konstanz, Juni 2019 Internationale Bevollmächtigtenkonferenz für die Bodenseefischerei (IBKF) IBKF – Internationale Bevollmächtigtenkonferenz für die Bodenseefischerei 2 Die Nase (Chondrostoma nasus) im Einzugsgebiet des Bodensees – Grundlagenbericht Die Nase (Chondrostoma nasus) im Einzugsgebiet des Bodensees Grundlagenbericht für internationale Maßnahmenprogramme Autor: Peter Rey GIS: John Hesselschwerdt Recherchen: Johannes Ortlepp Andreas Becker Begleitung: IBKF – Arbeitsgrupppe Wanderfische: Mag. DI Roland Jehle, Amt für Umwelt, Liechtenstein (Vorsitz) Dr. Marcel Michel, Amt für Jagd und Fischerei, Graubünden Roman Kistler, Jagd- und Fischereiverwalter des Kantons Thurgau Dario Moser, Jagd- und Fischereiverwalter des Kantons Thurgau LR Dr. Michael Schubert, Bayerische Landesanstalt für Landwirtschaft – Institut für Fischerei ORR Dr. Roland Rösch, Ministerium für Ländlichen Raum und Verbraucherschutz Baden-Württemberg Dr. Dominik Thiel, Amt für Natur, Jagd und Fischerei des Kantons St. Gallen Michael Kugler, Amt für Natur, Jagd und Fischerei des Kantons St. Gallen Mag. Nikolaus Schotzko, Amt der Vorarlberger Landesregierung, Landesfischereizentrum Vorarlberg RegD. Dr. Manuel Konrad, Regierungspräsidium Tübingen, Fischereibehörde Uwe Dußling, Regierungspräsidium Tübingen, Fischereibehörde Juni 2019 Internationale Bevollmächtigtenkonferenz -

EMS INFORMATION BULLETIN Nr 144
16/07/2021 EMSR517 – Flood in Western Germany EMSR518 – Flood in Belgium EMSR519 – Flood in Switzerland EMSR520 – Flood in The Netherlands EMS INFORMATION BULLETIN Nr 144 THE COPERNICUS EMERGENCY MANAGEMENT SERVICE The Copernicus Emergency Management Service forecasts, notifies, and monitors devastating floods in Germany, Netherlands, Belgium and Switzerland CEMS flood forecasting and notifying in Germany On 9 and 10 July, flood forecasts by the European Flood Awareness System (EFAS) of the Copernicus Emergency Management Service indicated a high probability of flooding for the Rhine River basin, affecting Switzerland and Germany. Subsequent forecasts also indicated a high risk of flooding for the Meuse River basin, affecting Belgium. The magnitude of the floods forecasted for the Rhine River basin increased significantly in this period. The first EFAS notifications were sent to the relevant national authorities starting on 10 July and, with the continuously updated forecasts, more than 25 notifications were sent for specific regions of the Rhine and Meuse River basins in the following days until 14 July. Figure: EFAS flood forecast from 12.07.2021 00:00 UTC Providing early and current maps of flooded areas On 13 July, the CEMS Rapid Mapping component was activated to map the ongoing floods in parts of Western Germany (EMSR517 Mapping Website , EMSR517 Activation Viewer). As a flood peak was foreseen on 16 July for segments of other rivers, CEMS preemptively acquired satellite images of the vulnerable area through Pre-Tasking on 14 July. These early images informed ensuing activations by the CEMS Rapid Mapping component based on the EFAS forecasts for areas in Belgium, Netherlands, Germany, Switzerland and France. -

Kids' Harbour Rally
Sommersstr. Yorckstr. Essener Münster- Barbarastr. Roßstr. Str. Schwerinstr. platz erst Paul-Spiegel- un r. Lützowstr. Gr Platz Zietenstr. Ahnfeldstr. Bankstr. Münsterstr. Jülicher Str. K ü h lw Brehmstr. Hombgr. Str. e Jülicher Str. tte Tußmannstr. rs Scharnhorststr. - tr. Seydlitzstr. Marc-Chagall-Str. Zietenstr. Bolten- Emmericher Str. Fischerstr. Düssel Cecilienallee sternstr. Collenbach Jordanstr. A T kämpchen o . n Mauerstr. r u t n Münsterstr. l Alt-Niederkassel Klever Str. Molkestr. s a o r s u str. Kolping- e t s Genger- l r . e S platz u c - r idt str. he Blücherstr. E A Stücker- Ahnfeldstr. l str. Schloßstr. l Speldorfer Str. e Am Deich e Schwerinstr. Klever Str. Pfalzstr. Weseler Str. Kaiserswerther Str. Kirchweg Kurt-Baurichter-Str. Robert-Lehr-Ufer Schorlemerstr . Beim r Dorf Niederkasseler Str. t Lennéstr. Peter-Roos-Str. Kanalstr. S r e d Habsbur- Fischerstr. l . Allee Toulouser gerstr. r e Pastor- t f s Mülheimer Str. u s na l Marc-Chagall Str. Brehm- Zentis-Weg e e Carl- Parkstr. k Ludwig- platz is n Mosterts- e i Wolker-Str. Augustastr. Nordstr. n W P Platz G . r r i Askanierstr. st n Schorlemerstr Leostr. ll K ha z a arsc - Perter- Johannsenstr. is Venloer Str. M G Derendorfer Str. - e e Lewitstr. Joachimstr. r o Stein Sittarder Str. - r Liebigstr. Rethelstr. F Paulus- g Stockkampstr. hauer- Moltkestr. r - i Weg e S Mönchenwerther Str. Tußmannstr. str. d Victoria- t Hartwich- r Achenbachstr. r N . Str. i platz Hohen- c Duisburger Str. e h t - t e R Franklinstr. staufenstr. Brüder- l i b n E.ON- Franklinstr. -

Ergebnisprotokoll Dialogforum 7
Ergebnisprotokoll, 7. Treffen am 26.09.2019 im Wissenschaftszentrum Bonn Dialogforum bonnbewegt. Hintergrund des Dialogforums Das Dialogforum bonnbewegt. ist ein wiederkehrendes Dialogformat. Es führt unterschiedliche Interessengruppen zum Thema Autobahnverkehr in Bonn zusammen. Vertreterinnen und Vertreter des ÖPNV, von Umweltverbänden, der regionalen Wirtschaft und Logistik, der Zivilgesellschaft sowie der Stadt Bonn und des Rhein-Sieg- Kreises sowie des Stadtmarketings und Tourismus, tauschen sich in diesem Rahmen mit dem Landesbetrieb Straßenbau NRW aus. Es werden Informationen zu den Hintergründen der jeweiligen Planungen, den anstehenden Baumaßnahmen und den Einschränkungen während der Bauzeit aus erster Hand vermittelt. Das Dialogforum bietet den Teilnehmenden die Möglichkeit sich über geplante Baumaßnahmen im Bonner Raum gegenseitig zu informieren, auszutauschen und zu vernetzen. Außerdem wird das Dialogforum genutzt, um die Entwicklung von Verkehrskonzepten zur Reduzierung der Belastungen und Umlenkung der Verkehrsströme zu diskutieren und anzustoßen. Folgende Institutionen sind zum Dialogforum eingeladen: ACE Bonn Haus & Grund Bonn/Rhein-Sieg e.V. ADAC Nordrhein IG BCE ADFC St. Augustin IHK Bonn/ Rhein-Sieg ADFC St. Beuel Knauber ADFC Niederkassel e.V. Kreishandwerkerschaft Bonn - Rhein Sieg Am Zehnhoff-Söns GmbH International Logistic M. Düren Transport GmbH & Co KG Services Metropolregien-Rheinland e.V. Bonner Hafenbetriebe GmbH NABU BUND Nahverkehr Rheinland (NVR) GmbH Bundesamt für Naturschutz Polizei Bonn Bundesministerium -

Von Der Quelle Zur Mündung, Eine Sedimentbilanz Des Rheins
Forschungs- und Entwicklungsprojekt der Bundesanstalt für Gewässerkunde im Rahmen der Ressortforschung des Bundesministeriums für Verkehr und digitale Infrastruktur Von der Quelle zur Mündung, eine Sedimentbilanz des Rheins Teil 3: Rheinnebenflüsse als Sedimentlieferanten BfG Bundesanstalt für Gewässerkunde IWW Lehrstuhl und Institut für Wasserbau und Wasserwirtschaft der RWTH Aachen University KHR Internationale Kommission für die Hydrologie des Rheingebietes BfG-1812 Bericht Von der Quelle zur Mündung, eine Sedimentbilanz des Rheins Rheinnebenflüsse als Sedimentlieferanten BfG-SAP-Nr. : M39610304039 Seitenzahl : 48 Bearbeitung : Nicole Gehres Birgit Astor Dr. Gudrun Hillebrand Koblenz, 17. September 2014 Der Bericht darf nur ungekürzt vervielfältigt werden. Die Vervielfältigung oder eine Veröffentlichungen bedürfen der schriftlichen Genehmigung der Bundesanstalt für Gewässerkunde. Bundesanstalt für Gewässerkunde Von der Quelle zur Mündung, eine Sedimentbilanz des Rheins Rheinnebenflüsse als Sediment- lieferanten Seite 2 Bundesanstalt für Gewässerkunde Von der Quelle zur Mündung, eine Sedimentbilanz des Rheins Inhaltsverzeichnis Rheinnebenflüsse als Sediment- lieferanten ................................................................................................................................................... 8 1. Hintergrund ......................................................................................................................... 9 2. Geographie, Hydrologie und Sedimentologie der Rheinnebenflüsse .......................... -

The Present Status of the River Rhine with Special Emphasis on Fisheries Development
121 THE PRESENT STATUS OF THE RIVER RHINE WITH SPECIAL EMPHASIS ON FISHERIES DEVELOPMENT T. Brenner 1 A.D. Buijse2 M. Lauff3 J.F. Luquet4 E. Staub5 1 Ministry of Environment and Forestry Rheinland-Pfalz, P.O. Box 3160, D-55021 Mainz, Germany 2 Institute for Inland Water Management and Waste Water Treatment RIZA, P.O. Box 17, NL 8200 AA Lelystad, The Netherlands 3 Administrations des Eaux et Forets, Boite Postale 2513, L 1025 Luxembourg 4 Conseil Supérieur de la Peche, 23, Rue des Garennes, F 57155 Marly, France 5 Swiss Agency for the Environment, Forests and Landscape, CH 3003 Bern, Switzerland ABSTRACT The Rhine basin (1 320 km, 225 000 km2) is shared by nine countries (Switzerland, Italy, Liechtenstein, Austria, Germany, France, Luxemburg, Belgium and the Netherlands) with a population of about 54 million people and provides drinking water to 20 million of them. The Rhine is navigable from the North Sea up to Basel in Switzerland Key words: Rhine, restoration, aquatic biodiversity, fish and is one of the most important international migration waterways in the world. 122 The present status of the river Rhine Floodplains were reclaimed as early as the and groundwater protection. Possibilities for the Middle Ages and in the eighteenth and nineteenth cen- restoration of the River Rhine are limited by the multi- tury the channel of the Rhine had been subjected to purpose use of the river for shipping, hydropower, drastic changes to improve navigation as well as the drinking water and agriculture. Further recovery is discharge of water, ice and sediment. From 1945 until hampered by the numerous hydropower stations that the early 1970s water pollution due to domestic and interfere with downstream fish migration, the poor industrial wastewater increased dramatically. -

Route Choices, Migration Speeds and Daily Migration Activity of European Silver Eels Anguilla Anguilla in the River Rhine, North-West Europe
Journal of Fish Biology (2009) 74, 2139–2157 doi:10.1111/j.1095-8649.2009.02293.x, available online at www.interscience.wiley.com Route choices, migration speeds and daily migration activity of European silver eels Anguilla anguilla in the River Rhine, north-west Europe A. W. Breukelaar*†, D. Ingendahl‡, F. T. Vriese§, G. de Laak¶, S. Staas** and J. G. P. Klein Breteler†† *Rijkswaterstaat Waterdienst, P. O. Box 17, 8200 AA Lelystad, The Netherlands, ‡Bezirksregierung Arnsberg, Heinsbergerstrasse 53, 57399 Kirchhundem-Albaum, Germany, §Visadvies bv, Twentehaven 5, 3433 PT Nieuwegein, The Netherlands, ¶Sportvisserij Nederland, Postbus 162, 3720 AD Bilthoven, The Netherlands, **Rheinfischereigenossenschaft im Lande Nordrhein-Westfalen, R¨omerhofweg 12, 50374 Erftstadt, Germany and ††Vivion bv, H¨andelstraat 18, 3533 GK Utrecht, The Netherlands Downstream migration of Anguilla anguilla silver eels was studied in the Lower Rhine, Germany, and the Rhine Delta, The Netherlands, in 2004–2006. Fish (n = 457) released near Cologne with implanted transponders were tracked by remote telemetry at 12 fixed detection locations distributed along the different possible migration routes to the North Sea. Relatively more A. anguilla migrated via the Waal than the Nederrijn, as would be expected from the ratio of river discharges at the bifurcation point at Pannerden. Downstream migration from the release site to Rhine-Xanten, close to the German–Dutch border, generally occurred in the autumn of the year of release but migration speeds tended to be low and variable and unaffected by maturation status or river discharge rates. Detection frequencies were not significantly related to discharge peaks or lunar cycles, but there was a minor detection peak 1–6 h after sunset. -
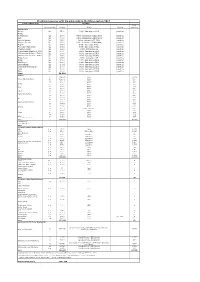
Stocking Measures with Big Salmonids in the Rhine System 2017
Stocking measures with big salmonids in the Rhine system 2017 Country/Water body Stocking smolt Kind and stage Number Origin Marking equivalent Switzerland Wiese Lp 3500 Petite Camargue B1K3 genetics Rhine Riehenteich Lp 1.000 Petite Camargue K1K2K4K4a genetics Birs Lp 4.000 Petite Camargue K1K2K4K4a genetics Arisdörferbach Lp 1.500 Petite Camargue F1 Wild genetics Hintere Frenke Lp 2.500 Petite Camargue K1K2K4K4a genetics Ergolz Lp 3.500 Petite Camargue K7C1 genetics Fluebach Harbotswil Lp 1.300 Petite Camargue K7C1 genetics Magdenerbach Lp 3.900 Petite Camargue K5 genetics Möhlinbach (Bachtele, Möhlin) Lp 600 Petite Camargue B7B8 genetics Möhlinbach (Möhlin / Zeiningen) Lp 2.000 Petite Camargue B7B8 genetics Möhlinbach (Zuzgen, Hellikon) Lp 3.500 Petite Camargue B7B8 genetics Etzgerbach Lp 4.500 Petite Camargue K5 genetics Rhine Lp 1.000 Petite Camargue B2K6 genetics Old Rhine Lp 2.500 Petite Camargue B2K6 genetics Bachtalbach Lp 1.000 Petite Camargue B2K6 genetics Inland canal Klingnau Lp 1.000 Petite Camargue B2K6 genetics Surb Lp 1.000 Petite Camargue B2K6 genetics Bünz Lp 1.000 Petite Camargue B2K6 genetics Sum 39.300 France L0 269.147 Allier 13457 Rhein (Alt-/Restrhein) L0 142.000 Rhine 7100 La 31.500 Rhine 3150 L0 5.000 Rhine 250 Doller La 21.900 Rhine 2190 L0 2.500 Rhine 125 Thur La 12.000 Rhine 1200 L0 2.500 Rhine 125 Lauch La 5.000 Rhine 500 Fecht und Zuflüsse L0 10.000 Rhine 500 La 39.000 Rhine 3900 L0 4.200 Rhine 210 Ill La 17.500 Rhine 1750 Giessen und Zuflüsse L0 10.000 Rhine 500 La 28.472 Rhine 2847 L0 10.500 Rhine 525 -

202.1017 Fld Riwa River Without
VISION FOR THE FUTURE THE QUALITY OF DRINKING WATER IN EUROPE MORE INFORMATION: How far are we? REQUIRES PREVENTIVE PROTECTION OF THE WATER Cooperation is the key word in the activities RESOURCES: of the Association of River Waterworks The Rhine (RIWA) and the International Association The most important of Waterworks in the Rhine Catchment aim of water pollution (IAWR). control is to enable the Cooperation is necessary to achieve waterworks in the structural, lasting solutions. Rhine river basin to Cooperation forges links between produce quality people and cultures. drinking water at any river time. Strong together without And that is one of the primary merits of this association of waterworks. Respect for the insights and efforts of the RIWA Associations of River other parties in this association is growing Waterworks borders because of the cooperation. This is a good basis for successful The high standards for IAWR International Association activities in coming years. the drinking water of Waterworks in the Rhine quality in Europe catchment area Some common activities of the associations ask for preventive of waterworks: protection of the water • a homgeneous international monitoring resources. Phone: +31 (0) 30 600 90 30 network in the Rhine basin, from the Alps Fax: +31 (0) 30 600 90 39 to the North Sea, • scientific studies on substance and parasites Priority must be given E-mail: [email protected] which are relevant for to protecting water [email protected] drinking water production, resources against publication of monitoring pollutants that can The prevention of Internet: www.riwa.org • results and scientific studie get into the drinking water pollution www.iawr.org in annual reports and other water supply. -

Focus 5 Nachtigallenweg 39 · 53844 Troisdorf (D)
© J. Fischer Fischer © J. The Rhine One river basin - many visitors‘ centres 1 Afsluitdijk Wadden Center Sluisweg 1a · 8752 TR Kornwerderzand (NL) © IKSR www.theafsluitdijk.com 2 Bird observatory - near the Haringvliet dam Scheelhoek Stellendam (NL) www.vogelbescherming.nl ICPR headquarters in Koblenz www.haringvliet.nu 3 Wissenshaus Wanderfische Wahnbachtalstraße 13a · 53721 Siegburg (D) http://www.wasserlauf-nrw.de/en/ 4 Wildlachszentrum Rhein-Sieg Wahnbachtalstraße 13a · 53721 Siegburg (D) http://www.wasserlauf-nrw.de/en/ Fischereimuseum Bergheim an der Sieg Focus 5 Nachtigallenweg 39 · 53844 Troisdorf (D) www.fischereimuseum-bergheim-sieg.de 6 Zoological Research Museum Alexander König incl. permanent exhibition Rhine „FRESHWATER - Life in Flow “ Adenauerallee 160 · 53113 Bonn (D) Further information What can be experienced and where? https://www.zfmk.de/en/museum/ Visitors’ centres in the Rhine catchment permanent-exhibitions/new-freshwater International Commission for the Protection of the Rhine (ICPR) 7 Mosellum Postfach 20 02 53 Peter-Altmeier-Ufer 1 · 56068 Koblenz (D) D-56002 Koblenz www.mosellum.rlp.de Tel: 0049-(0)261-94252-0 Lahnfenster Hessen 8 Fax: 0049-(0)261-94252-52 Bootshausstraße 8 · 35390 Gießen (D) www.rp-giessen.hessen.de/ [email protected] umwelt-natur/landwirtschaft-fischerei/ www.iksr.org lahnfenster-hessen © IKSR-CIPR-ICBR-ICPR 2017 © J. Schneider Wikimedia cc 9 Biodiversum 5, Bréicherwee · 5441 Remerschen (LU) Experience biological diversity and water protection www.environnement.public.lu/ conserv_nature/Centres_d_accueil/ …at several places in the Rhine basin between Switzerland Due to a multitude of obstacles, numerous spawning and CA_Biodiversum/index.html and the North Sea coast! juvenile waters in the tributaries are today still not accessible The offer is varied and comprises nature protection centres, or only accessible to a very limited extent. -

Einladung Zu Einer Lesung
Nr. 13 Donnerstag, 31. März 2016 Einladung zu einer Lesung am 05. April 2016, 19.30 Uhr mit Bov Bjerg Donnerstag, 31.03.2016 14.30 Uhr Seniorentreff „Auerhaus“ Samstag, 02.04.2016 ab 14.30 Uhr Handballspiele Sonntag, 03.04.2016 Unter dem Motto „Lebenswege und Kurswechsel“ findet das deutsch- ab 12.45 Uhr Handballspiele schweizerische Literaturfestival „Erzählzeit ohne Grenzen“ Singen- Montag, 04.04.2016 Schaffhausen vom 02. bis 10. April 2016 statt. Es bietet erneut span- 12.00 Uhr Bürger für Bürger Mittagessen nende Begegnungen an außergewöhnlichen Leseorten in der Region 19.30 Uhr Bibelabend im Remigiushaus zwischen Bodensee und Rheinfall. Dienstag, 05.04.2016 14.30 Uhr Ev. Kirche Seniorennachmittag In der Mensa der Gemeinschaftsschule (Gebäude C), Gartenstraße 2, 19.30 Uhr Erzählzeit ohne Grenzen / 78256 Steißlingen wird am Dienstag, 05. April 2016, 19.30 Uhr der Au- Lesung tor Bov Bjerg aus seinem Roman „Auerhaus“ lesen. Der Eintritt ist frei. Mittwoch, 06.04.2016 10.00-12.00 Uhr Bürger für Bürger Im „Auerhaus“, einem leer stehenden alten Bauernhaus finden sich Anfang Büro geöffnet der 1980er-Jahre einige Jugendliche eines katholisch geprägten Dorfes zu 12.00 Uhr Bürger für Bürger Mittagessen 14.00-18.00 Uhr Schulanmeldungen einer Schüler-WG zusammen. Sie kümmern sich um einen Freund, der see- neue 1. Klässler lisch am Ende ist und suchen nach ihren Wegen zum Glück. Von Eltern und 18.00 Uhr Tourist Information der Dorfgemeinschaft geduldet, gestalten sie mit Einfallsreichtum ihren All- Vermietertreff 19.00 Uhr Informationsabend tag zwischen Althergebrachtem und Gesetzlosigkeit. Solarstromerzeugung In seinem mitreißenden Bestseller-Roman erzählt Bov Bjerg von Liebe und Donnerstag, 07.04.2016 14.30 Uhr Seniorentreff Freundschaft und von jungen Leuten, die auf unterschiedliche Weise ihr Glück suchen. -

Template for Creating Reports in Delft Hydraulics Style V.3.01
Draft Prepared for: RIZA Arnhem FEWS-Rhine version 1.02 Improvements and adjustments Report November 2004 Q3618.00 Prepared for: RIZA Arnhem FEWS-Rhine version 1.02 Improvements and adjustments Albrecht Weerts & Hanneke Van de Klis Report November, 2004 FEWS-Rhine version 1.02 Q3618.00 November 2004 Improvements and adjustments Contents 1 Introduction.........................................................................................................1—1 2 State Management SOBEK................................................................................2—1 2.1 Introduction..............................................................................................2—1 2.2 State handling FEWS general ..................................................................2—1 2.3 SOBEK ....................................................................................................2—2 3 Implementation of a new SOBEK model..........................................................3—1 3.1 Introduction..............................................................................................3—1 3.2 Important file ...........................................................................................3—1 3.3 Implementation new SOBEK model .......................................................3—2 3.4 Implementation new SOBEK executable ................................................3—2 3.5 Implementation adapted SOBEK model..................................................3—2 3.6 Implementation of an extra SOBEK model.............................................3—3