Draft Genome Assembly of the Freshwater Apex Predator Wels Catfish (Silurus Glanis) Using Linked-Read Sequencing Mikhail Yu
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Zootaxa,Taxonomy of the Catfish Genus Pseudoplatystoma Bleeker
Zootaxa 1512: 1–38 (2007) ISSN 1175-5326 (print edition) www.mapress.com/zootaxa/ ZOOTAXA Copyright © 2007 · Magnolia Press ISSN 1175-5334 (online edition) Taxonomy of the catfish genus Pseudoplatystoma Bleeker (Siluriformes: Pimelodidae) with recognition of eight species URIEL ANGEL BUITRAGO–SUÁREZ and BROOKS M. BURR Department of Biological Sciences, Harper College, Palatine, Illinois 60067, USA; Telephone: 847–925–6718, e–mail: [email protected] and Department of Zoology, Southern Illinois University Carbondale, Carbondale, Illinois 62901, USA Abstract The genus Pseudoplatystoma Bleeker consists of three species long recognized as: P. fasciatum (Linnaeus), P. tigrinum (Valenciennes), and P. corruscans (Spix & Agassiz), and five species recently recognized or described here: P. punctifer (Castelnau), P. reticulatum Eigenmann & Eigenmann, P. orinocoense n. sp., P. m et ae n se n. sp., and P. magdaleniatum n. sp. The eight species form a monophyletic group with two clades that are supported by anatomical features (i.e., skeletal anatomy and myology). One clade (P. tigrinum and P. m e ta e ns e) is restricted to the Orinoco and Amazon basins, and the other clade, comprised of the remaining six species, is found in the Guyanas, Orinoco, Amazon, and Paraná basins. The species are diagnosed on the basis of body shape, color pattern (e.g., bars, loops, and spots), skeletal anatomy, and verte- bral numbers. Pseudoplatystoma punctifer and P. tigrinum) are sympatric in the Amazon Basin, P. m e ta e ns e and P. orinocoense in the Orinoco Basin, and P. corruscans and P. reticulatum, are sympatric in the Paraná. Pseudoplatystoma magdaleniatum (Magdalena basin) and P. -

Amur Fish: Wealth and Crisis
Amur Fish: Wealth and Crisis ББК 28.693.32 Н 74 Amur Fish: Wealth and Crisis ISBN 5-98137-006-8 Authors: German Novomodny, Petr Sharov, Sergei Zolotukhin Translators: Sibyl Diver, Petr Sharov Editors: Xanthippe Augerot, Dave Martin, Petr Sharov Maps: Petr Sharov Photographs: German Novomodny, Sergei Zolotukhin Cover photographs: Petr Sharov, Igor Uchuev Design: Aleksey Ognev, Vladislav Sereda Reviewed by: Nikolai Romanov, Anatoly Semenchenko Published in 2004 by WWF RFE, Vladivostok, Russia Printed by: Publishing house Apelsin Co. Ltd. Any full or partial reproduction of this publication must include the title and give credit to the above-mentioned publisher as the copyright holder. No photographs from this publication may be reproduced without prior authorization from WWF Russia or authors of the photographs. © WWF, 2004 All rights reserved Distributed for free, no selling allowed Contents Introduction....................................................................................................................................... 5 Amur Fish Diversity and Research History ............................................................................. 6 Species Listed In Red Data Book of Russia ......................................................................... 13 Yellowcheek ................................................................................................................................... 13 Black Carp (Amur) ...................................................................................................................... -

Defining Habitat Demands of Wels Catfish (Silurus Glanis) in a Swedish Lake - a Look Into Muddy Waters
Faculty of Natural Resources and Agricultural Sciences Defining habitat demands of Wels catfish (Silurus glanis) in a Swedish lake - A look into muddy waters David Spange Department of Aquatic Resources Institute of Freshwater Research Master’s thesis • 30 hec Master Programme in Biology - Limnology Drottningholm 2018 Defining habitat demands of Wels catfish (Silurus glanis) in a Swedish lake - A look into muddy waters Definiering av den europeiska malens (Silurus glanis) habitatkrav i en svensk sjö. David Spange Supervisor: Henrik Jeuthe, Swedish University of Agricultural Sciences, Department of aquatic resources Assistant supervisor: Joep De Leeuw, Swedish University of Agricultural Sciences, Department of aquatic resources Examiner: Kerstin Holmgren, Swedish University of Agricultural Sciences, Department of aquatic resources Credits: 30 hec Level: A2E Course title: Independent Project in Biology – Master’s thesis Course code: EX0565 Programme/education: Master Programme in Biology Limnology – Ecology and Environment of Inland Waters, 120 hec Place of publication: Drottningholm Year of publication: 2018 Cover picture: David spange Online publication: https://stud.epsilon.slu.se Keywords: Wels catfish, Silurus glanis, Sweden, Spawning, Spawning habitats, acoustic camera, ARIS, telemetry Sveriges lantbruksuniversitet Swedish University of Agricultural Sciences Faculty of Natural Resources and Agricultural Sciences Department of Aquatic Resources Abstract The wels catfish (Silurus glanis) is a rare species in Swedish waters, it demands higher water temperatures than most lakes and rivers can offer. One of the few locations with naturally occurring wels catfish in Sweden is Lake Båven, situated about an hour and a half south of Stockholm, in the county of Södermanland. Due to declines of the Swedish catfish populations, which are mostly a result of human impacts such as the destruction of spawning habitats, conservational actions are now needed in purpose to secure the future of the species in Sweden. -
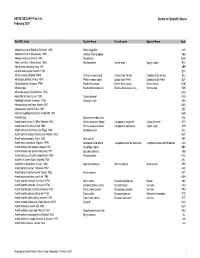
ASFIS ISSCAAP Fish List February 2007 Sorted on Scientific Name
ASFIS ISSCAAP Fish List Sorted on Scientific Name February 2007 Scientific name English Name French name Spanish Name Code Abalistes stellaris (Bloch & Schneider 1801) Starry triggerfish AJS Abbottina rivularis (Basilewsky 1855) Chinese false gudgeon ABB Ablabys binotatus (Peters 1855) Redskinfish ABW Ablennes hians (Valenciennes 1846) Flat needlefish Orphie plate Agujón sable BAF Aborichthys elongatus Hora 1921 ABE Abralia andamanika Goodrich 1898 BLK Abralia veranyi (Rüppell 1844) Verany's enope squid Encornet de Verany Enoploluria de Verany BLJ Abraliopsis pfefferi (Verany 1837) Pfeffer's enope squid Encornet de Pfeffer Enoploluria de Pfeffer BJF Abramis brama (Linnaeus 1758) Freshwater bream Brème d'eau douce Brema común FBM Abramis spp Freshwater breams nei Brèmes d'eau douce nca Bremas nep FBR Abramites eques (Steindachner 1878) ABQ Abudefduf luridus (Cuvier 1830) Canary damsel AUU Abudefduf saxatilis (Linnaeus 1758) Sergeant-major ABU Abyssobrotula galatheae Nielsen 1977 OAG Abyssocottus elochini Taliev 1955 AEZ Abythites lepidogenys (Smith & Radcliffe 1913) AHD Acanella spp Branched bamboo coral KQL Acanthacaris caeca (A. Milne Edwards 1881) Atlantic deep-sea lobster Langoustine arganelle Cigala de fondo NTK Acanthacaris tenuimana Bate 1888 Prickly deep-sea lobster Langoustine spinuleuse Cigala raspa NHI Acanthalburnus microlepis (De Filippi 1861) Blackbrow bleak AHL Acanthaphritis barbata (Okamura & Kishida 1963) NHT Acantharchus pomotis (Baird 1855) Mud sunfish AKP Acanthaxius caespitosa (Squires 1979) Deepwater mud lobster Langouste -

Growth and Variation in Eurypterus Remipes Dekay
Bull. geo!. Instn Univ. Upsala: N. S. 4, 6: 81-114, 1974 GROWTH AND VARIATION IN EURYPTERUS REMIPES DEKAY By H. E. Andrews, ]. C. Brower, S. ]. Gould, and R. A. Reyment Andrews, H. E., Brower, ]. C., Gould, S. ]., Reyment, R. A.: Growth and Variation in EurypteruJ remipes DeKay, Bull. geo!. Instn Univ. Upsala, Vol. N. S 4, pp. 81-114. Th e statistic al analysis of two subspecies of Eurypterus remipes shows that both of them display very high integratio:� among all measures considered. The prosomal set of variables are highly integrated with the body set. There is little allometry in bivariate growth sequences. As best known at the present time, trilobites show an analogous leve! of integration; there is therefore reason to suspect that the gr owth relationships here recorded are wide-spread among some arthropods. Ontogenetic growth is analysed for E. remipes remipes, after the establishment of growth stages by a stepwise multivariate technique. Canonical correlation is used to examine the pattern of integration between head and body. This is, again, exceptionally high. Other methods of multi variate statistics are applied the analysis of the underlying relationships between variables. The palaeoec ology of the Fiddlersto Gr een Member (Bertie Formation, Upper Silurian) is discussed. H. E. Andrews, Department of Geology, \'(/ellesley Colle�;e, \'(/ellesley, Mass., U.S.A., S. ]. Gould, Museum of Comparative Zoology, Harvard University, Cambridge, Mass., U.S.A., ]. C. Brower, Department of Geology, Syracuse University, U.S.A., R. A. Reyment, Paleontologiska Institutionen, Uppsala Unit;ersitet. Su,eden. INTRODUCTION remipes and the E. tetragonophthalmus of Fischer Ever smce Mitchill (1818) recorded the first de Waldheim (synonym E. -
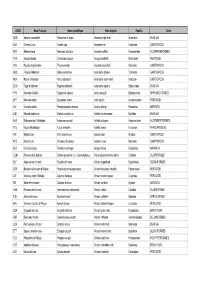
Liste Espèces
CODE Nom Français Nom scientifique Nom Anglais Famille Ordre ODQ Anomie cascabelle Pododesmus cepio Abalone jingle shell Anomiidae BIVALVIA ABX Ormeaux nca Haliotis spp Abalones nei Haliotidae GASTROPODA REN Sébaste rose Sebastes fasciatus Acadian redfish Scorpaenidae SCORPAENIFORMES YNA Acoupa toeroe Cynoscion acoupa Acoupa weakfish Sciaenidae PERCOIDEI HSV Pourpre aiguillonnee Thais aculeata Aculeate rock shell Muricidae GASTROPODA GBQ Troque d'Adanson Gibbula adansoni Adanson's gibbula Trochidae GASTROPODA NKA Natice d'Adanson Natica adansoni Adanson's moon snail Naticidae GASTROPODA GLW Tagal d'Adanson Tagelus adansonii Adanson's tagelus Solecurtidae BIVALVIA PYD Manchot d'Adélie Pygoscelis adeliae Adelie penguin Spheniscidae SPHENISCIFORMES QFT Maconde aden Synagrops adeni Aden splitfin Acropomatidae PERCOIDEI NIV Crevette adonis Parapenaeopsis venusta Adonis shrimp Penaeidae NATANTIA DJD Modiole adriatique Modiolus adriaticus Adriatic horse mussel Mytilidae BIVALVIA AAA Esturgeon de l'Adriatique Acipenser naccarii Adriatic sturgeon Acipenseridae ACIPENSERIFORMES FCV Fucus d'Adriatique Fucus virsoides Adriatic wrack Fucaceae PHAEOPHYCEAE IRR Mitre brûlée Mitra eremitarum Adusta miter Mitridae GASTROPODA KCE Murex bruni Chicoreus brunneus Adusta murex Muricidae GASTROPODA AES Crevette ésope Pandalus montagui Aesop shrimp Pandalidae NATANTIA CGM Poisson-chat, hybride Clarias gariepinus x C. macrocephalus Africa-bighead catfish, hybrid Clariidae SILURIFORMES SUF Ange de mer africain Squatina africana African angelshark Squatinidae SQUALIFORMES -

Siluriformes: Ariidae), from the Atlantic Coast of South America, with Insights Into the Demography and Biogeography of the Species
Neotropical Ichthyology, 14(2): e150184, 2016 Journal homepage: www.scielo.br/ni DOI: 10.1590/1982-0224-20150184 Published online: 27 June 2016 (ISSN 1982-0224) Early Pleistocene lineages of Bagre bagre (Linnaeus, 1766) (Siluriformes: Ariidae), from the Atlantic coast of South America, with insights into the demography and biogeography of the species Wemerson C. da Silva1, Alexandre P. Marceniuk1,2, João Braullio L. Sales1,3 and Juliana Araripe1 Coastal and marine environments are characterized by a lack of evident physical barriers or geographic isolation, and it may be difficult to understand how divergence can arise and be sustained in marine environments. The identification of ‘soft’ barriers is a crucial step towards the understanding of gene flow in marine environments. The marine catfishes of the family Ariidae are a demersal group with restricted migratory behavior, no pelagic larval stages, and mechanisms of larval retention, representing a potentially useful model for the understanding of historical processes of allopatric speciation in the marine environment. In the present study, two lineages of the Coco sea catfish, Bagre bagre, were recognized from their complete segregation at both mitochondrial and morphological levels. One lineage is distributed between Venezuela and the northern coast of Brazil, including the semiarid northeast coast, while the second lineage is found on the eastern coast of Brazil, including the humid northeast coast. Based on distribution area, habitats preference, and genetic variability, inferences are made in relation to biogeography and demography of lineages in Atlantic coast of South America. Ambientes marinhos costeiros são caracterizados pela falta de barreiras físicas evidentes e ausência de isolamento geográfico, sendo difícil compreender como diferentes linhagens podem surgir e ser mantidas nestes ambientes. -

Silurus Glanis in Its Native and Introduced Ranges*
F I S H and F I S H E R I E S , 2009, 10, 252–282 Voracious invader or benign feline? A review of the environmental biology of European catfish Silurus glanis in its native and introduced ranges* Gordon H Copp1,2, J Robert Britton2, Julien Cucherousset2,3, Emili Garcı´a-Berthou4, Ruth Kirk5, Edmund Peeler6 & Saulius Stakenas_ 7 1Salmon & Freshwater Fisheries Team, Centre for Environment, Fisheries & Aquaculture Science, Pakefield Road, Lowestoft, Suffolk NR33 0HT, UK; 2Centre for Conservation Ecology, School of Conservation Sciences, Bournemouth University, Fern Barrow, Poole, Dorset, BH12 5BB, UK; 3EcoLab Laboratoire d’E´cologie Fonctionnelle, UMR 5245 (CNRS- UPS-INPT), Universite´ Paul Sabatier, Baˆt. 4R3, 118, route de Narbonne, F-31062 Toulouse, Cedex 9, France; 4Institute of Aquatic Ecology, University of Girona, E-17071 Girona, Spain; 5School of Life Sciences, Kingston University, Penrhyn Road, Kingston upon Thames, Surrey KT1 2EE, UK; 6Centre for Environment, Fisheries and Aquaculture Science, Weymouth, Dorset DT4 8UB, UK; 7Department of Freshwater Ecology, Institute of Ecology of Vilnius University, Akademijos 2, LT-08412, Vilnius, Lithuania Abstract Correspondence: A popular species for food and sport, the European catfish (Silurus glanis) is well-studied Gordon H Copp, Salmon & Freshwater in its native range, but little studied in its introduced range. Silurus glanis is the largest- Fisheries Team, Cen- bodied freshwater fish of Europe and is historically known to take a wide range of food tre for Environment, items including human remains. As a result of its piscivorous diet, S. glanis is assumed Fisheries & Aquacul- to be an invasive fish species presenting a risk to native species and ecosystems. -
A Review of the Catfish Genus Pterocryptis (Siluridae)
Journal of Fish Biology (2001) 59, 624–644 doi:10.1006/jfbi.2001.1673, available online at http://www.idealibrary.com on A review of the catfish genus Pterocryptis (Siluridae) in Vietnam, with the description of two new species H. H. N*†¶ J. F‡ *Fish Division, Museum of Zoology, University of Michigan, 1109 Geddes Avenue, Ann Arbor, Michigan 48109-1079, U.S.A., †Department of Biological Sciences, National University of Singapore, 10 Kent Ridge Crescent, Singapore 119260 and ‡Institut fu¨r Gewa¨ssero¨kologie und Binnenfischerei, Mu¨ggelseedamm 310, D-12561 Berlin, Germany (Received 9 December 2000, Accepted 23 May 2001) Of the silurid catfish genus Pterocryptis in Vietnam, a total of four species are recognized as valid, of which Pterocryptis crenula and P. verecunda are described here as new. The type locality of P. cochinchinensis is restricted to central Vietnam, and the species is redescribed from topotypic material. Pterocryptis crenula (from northeastern Vietnam) can be differentiated from its congeners in having visibly confluent anal and caudal fins while P. verecunda (from Cat Ba Island in northeastern Vietnam) can be differentiated from its congeners in having the genital papilla concealed behind the anus. Notes on the identity of other nominal Pterocryptis species are also provided. 2001 The Fisheries Society of the British Isles Key words: new species; taxonomy; Siluridae; Pterocryptis. INTRODUCTION Silurid catfishes of the genus Pterocryptis Peters, 1861, are usually found in relatively fast-flowing mountain streams throughout India, southern China and South-east Asia. The genus was recently removed from synonymy with Silurus Linnaeus, 1758, by Bornbusch (1991), and the taxonomy of many of its species has not been fully resolved. -
Wels Catfish Silurus Glanis
Wels catfish Silurus glanis Description Identification Scaleless, elongated body. It can grow up to 13 feet long with a weight of over 880 pounds. Upper side is usually a dark color and the flanks and belly are more pale. Fins are brownish and the body has a mottled appearance that is sometimes accompanied by brown spots. 1 dorsal spone and 4-5 dorsal soft rays, 1 anal spine and 90-94 anal soft rays and a caudal fin with 17 rays. Habitat Native to central, southern and easter Europe and near the Baltic and Caspian Seas. It prefers large, warm lakes and deep, slow-flowing rivers Source: MISIN. 2021. Midwest Invasive Species Information Network. Michigan State University - Applied Spatial Ecology and Technical Services Laboratory. Available online at https://www.misin.msu.edu/facts/detail.php?id=273. where it can remain sheltered in holes, sunken trees, etc. Reproduction Breed annually during spring; hatching takes 3-10 days; reproductive maturity is 4 years for a female and 3 years for a male. The male creates a shallow depression that will hold thousands of eggs. Females can lay up to 30,000 eggs per kilogram of body weight. Impact Have been implicated in declining populations of other commerical fishes. Capable of carrying bacterial disease that can be transmitted to other fish such as Red head disease (Vibrio sp. bacterium) and Gill disease (Flavobacterium). Similar Aristotle's catfish (Silurus aristotelis); Amur catfish (Silurus asotus); Giant lake biwa catfish (Silurus biwaensis); Soldatov's catfish (Silurus soldatovi); Mekong giant catfish (Pangasianodon gigas). Monitoring and Rapid Response Credits The information provided in this factsheet was gathered from the Wikipedia.Individual species images that appear with a number in a black box are courtesy of the Bugwood.org network Source: MISIN. -

Ting-Bing ZHU, Jun HUANG, Xu-Ge WANG, and De-Guo YANG*
ACTA ICHTHYOLOGICA ET PISCATORIA (2018) 48 (2): 205–207 DOI: 10.3750/AIEP/02394 FIRST RECORD OF EXOTIC AMUR CATFISH, SILURUS ASOTUS (ACTINOPTERYGII: SILURIFORMES: SILURIDAE), IN THE TIBET STRETCH OF THE LANCANG RIVER, CHINA Ting-Bing ZHU, Jun HUANG, Xu-Ge WANG, and De-Guo YANG* Key Laboratory of Freshwater Biodiversity Conservation, Ministry of Agriculture of China, Yangtze River Fisheries Research Institute, Chinese Academy of Fisheries Science, Wuhan, China Zhu T.-B., Huang J., Wang X.-G., Yang D.-G. 2018. First record of exotic Amur catfish, Silurus asotus (Actinopterygii: Siluriformes: Siluridae), in the Tibet stretch of the Lancang River, China. Acta Ichthyol. Piscat. 48 (2): 205–207. Abstract. A single specimen of exotic Amur catfish, Silurus asotus Linnaeus, 1758, was captured by gillnet in the Tibet stretch of the Lancang River for the first time. (The Lancang River is an international river and in its lower course is also known as the Mekong River.) It is the first record of this fish species in eastern Tibet. The most possible pathway of the Amur catfish introduction into the Tibet stretch of the Lancang River was a human- mediated release known as life liberation activities or life release ceremony. Keywords: Silurus asotus, exotic fish, the Lancang River, Mekong River, Tibet, life release ceremony INTRODUCTION MATERIAL AND METHODS The Lancang River is a big international river in Asia. A single specimen of Amur catfish was sampled The Lancang River in its lower course is also known as (Fig. 2) using gillnet (length 10 m, height 1.5 m, mesh the Mekong River. -

Review Article Review of the Freshwater Catfishes Of
Iran. J. Ichthyol. (December 2014), 1(4): 218–257 Received: November 15, 2014 © 2014 Iranian Society of Ichthyology Accepted: December 02, 2014 P-ISSN: 2383-1561; E-ISSN: 2383-0964 doi: http://www.ichthyol.ir Review Article Review of the Freshwater Catfishes of Iran (Order Siluriformes) Brian W. COAD Canadian Museum of Nature, Ottawa, Ontario, K1P 6P4 Canada. Email: [email protected] Abstract: The systematics, morphology, distribution, biology, economic importance and conservation of the freshwater catfishes of Iran are described, the species are illustrated, and a bibliography on these fishes is provided. There are four families with four genera and six species, one of which is an exotic; the family Bagridae with one genus and species, Mystus pelusius in the Persian Gulf, Hormuz and Tigris River basins; the family Heteropneustidae with one genus and species, Heteropneustes fossilis in the Tigris River basin; the family Siluridae with one genus and two species, Silurus glanis in the Caspian Sea, Lake Orumiyeh and Hari River basins and Silurus triostegus in the Tigris River basin; and the family Sisoridae with one genus and two species, Glyptothorax kurdistanicus in the Tigris River basin and Glyptothorax silviae in the Persian Gulf and Tigris River basins. There are also some species in the neighboring regions which were not reported from Iran. Keywords: Bagridae, Biology, Heteropneustidae, Morphology, Siluridae, Sisoridae. Introduction (1996) and Coad (1998) and their familial and The freshwater ichthyofauna of Iran comprises a generic relationships with neighbouring basins in diverse set of families and species. These form other countries in Coad (1996a). important elements of the aquatic ecosystem and a These fishes range in size from under 10cm to number of species are of commercial or other over 3m, one of the largest species, Silurus glanis, significance.