Identification of Genes with a Correlation Between Copy Number
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Analysis of Trans Esnps Infers Regulatory Network Architecture
Analysis of trans eSNPs infers regulatory network architecture Anat Kreimer Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Graduate School of Arts and Sciences COLUMBIA UNIVERSITY 2014 © 2014 Anat Kreimer All rights reserved ABSTRACT Analysis of trans eSNPs infers regulatory network architecture Anat Kreimer eSNPs are genetic variants associated with transcript expression levels. The characteristics of such variants highlight their importance and present a unique opportunity for studying gene regulation. eSNPs affect most genes and their cell type specificity can shed light on different processes that are activated in each cell. They can identify functional variants by connecting SNPs that are implicated in disease to a molecular mechanism. Examining eSNPs that are associated with distal genes can provide insights regarding the inference of regulatory networks but also presents challenges due to the high statistical burden of multiple testing. Such association studies allow: simultaneous investigation of many gene expression phenotypes without assuming any prior knowledge and identification of unknown regulators of gene expression while uncovering directionality. This thesis will focus on such distal eSNPs to map regulatory interactions between different loci and expose the architecture of the regulatory network defined by such interactions. We develop novel computational approaches and apply them to genetics-genomics data in human. We go beyond pairwise interactions to define network motifs, including regulatory modules and bi-fan structures, showing them to be prevalent in real data and exposing distinct attributes of such arrangements. We project eSNP associations onto a protein-protein interaction network to expose topological properties of eSNPs and their targets and highlight different modes of distal regulation. -

ACE2 Interaction Networks in COVID-19: a Physiological Framework for Prediction of Outcome in Patients with Cardiovascular Risk Factors
Journal of Clinical Medicine Article ACE2 Interaction Networks in COVID-19: A Physiological Framework for Prediction of Outcome in Patients with Cardiovascular Risk Factors Zofia Wicik 1,2 , Ceren Eyileten 2, Daniel Jakubik 2,Sérgio N. Simões 3, David C. Martins Jr. 1, Rodrigo Pavão 1, Jolanta M. Siller-Matula 2,4,* and Marek Postula 2 1 Centro de Matemática, Computação e Cognição, Universidade Federal do ABC, Santo Andre 09606-045, Brazil; zofi[email protected] (Z.W.); [email protected] (D.C.M.J.); [email protected] (R.P.) 2 Department of Experimental and Clinical Pharmacology, Medical University of Warsaw, Center for Preclinical Research and Technology CEPT, 02-091 Warsaw, Poland; [email protected] (C.E.); [email protected] (D.J.); [email protected] (M.P.) 3 Federal Institute of Education, Science and Technology of Espírito Santo, Serra, Espírito Santo 29056-264, Brazil; [email protected] 4 Department of Internal Medicine II, Division of Cardiology, Medical University of Vienna, 1090 Vienna, Austria * Correspondence: [email protected]; Tel.: +43-1-40400-46140; Fax: +43-1-40400-42160 Received: 9 October 2020; Accepted: 17 November 2020; Published: 21 November 2020 Abstract: Background: Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection (coronavirus disease 2019; COVID-19) is associated with adverse outcomes in patients with cardiovascular disease (CVD). The aim of the study was to characterize the interaction between SARS-CoV-2 and Angiotensin-Converting Enzyme 2 (ACE2) functional networks with a focus on CVD. Methods: Using the network medicine approach and publicly available datasets, we investigated ACE2 tissue expression and described ACE2 interaction networks that could be affected by SARS-CoV-2 infection in the heart, lungs and nervous system. -

Table 2. Significant
Table 2. Significant (Q < 0.05 and |d | > 0.5) transcripts from the meta-analysis Gene Chr Mb Gene Name Affy ProbeSet cDNA_IDs d HAP/LAP d HAP/LAP d d IS Average d Ztest P values Q-value Symbol ID (study #5) 1 2 STS B2m 2 122 beta-2 microglobulin 1452428_a_at AI848245 1.75334941 4 3.2 4 3.2316485 1.07398E-09 5.69E-08 Man2b1 8 84.4 mannosidase 2, alpha B1 1416340_a_at H4049B01 3.75722111 3.87309653 2.1 1.6 2.84852656 5.32443E-07 1.58E-05 1110032A03Rik 9 50.9 RIKEN cDNA 1110032A03 gene 1417211_a_at H4035E05 4 1.66015788 4 1.7 2.82772795 2.94266E-05 0.000527 NA 9 48.5 --- 1456111_at 3.43701477 1.85785922 4 2 2.8237185 9.97969E-08 3.48E-06 Scn4b 9 45.3 Sodium channel, type IV, beta 1434008_at AI844796 3.79536664 1.63774235 3.3 2.3 2.75319499 1.48057E-08 6.21E-07 polypeptide Gadd45gip1 8 84.1 RIKEN cDNA 2310040G17 gene 1417619_at 4 3.38875643 1.4 2 2.69163229 8.84279E-06 0.0001904 BC056474 15 12.1 Mus musculus cDNA clone 1424117_at H3030A06 3.95752801 2.42838452 1.9 2.2 2.62132809 1.3344E-08 5.66E-07 MGC:67360 IMAGE:6823629, complete cds NA 4 153 guanine nucleotide binding protein, 1454696_at -3.46081884 -4 -1.3 -1.6 -2.6026947 8.58458E-05 0.0012617 beta 1 Gnb1 4 153 guanine nucleotide binding protein, 1417432_a_at H3094D02 -3.13334396 -4 -1.6 -1.7 -2.5946297 1.04542E-05 0.0002202 beta 1 Gadd45gip1 8 84.1 RAD23a homolog (S. -

Datasheet: VPA00586KT Product Details
Datasheet: VPA00586KT Description: EIF3E ANTIBODY WITH CONTROL LYSATE Specificity: EIF3E Format: Purified Product Type: PrecisionAb™ Polyclonal Isotype: Polyclonal IgG Quantity: 2 Westerns Product Details Applications This product has been reported to work in the following applications. This information is derived from testing within our laboratories, peer-reviewed publications or personal communications from the originators. Please refer to references indicated for further information. For general protocol recommendations, please visit www.bio-rad-antibodies.com/protocols. Yes No Not Determined Suggested Dilution Western Blotting 1/1000 PrecisionAb antibodies have been extensively validated for the western blot application. The antibody has been validated at the suggested dilution. Where this product has not been tested for use in a particular technique this does not necessarily exclude its use in such procedures. Further optimization may be required dependant on sample type. Target Species Human Species Cross Reacts with: Rat Reactivity N.B. Antibody reactivity and working conditions may vary between species. Product Form Purified IgG - liquid Preparation 20μl Rabbit polyclonal antibody purified by affinity chromatography Buffer Solution Phosphate buffered saline Preservative 0.09% Sodium Azide (NaN3) Stabilisers 2% Sucrose Immunogen Synthetic peptide directed towards the middle region of human EIF3E External Database Links UniProt: P60228 Related reagents Entrez Gene: 3646 EIF3E Related reagents Synonyms EIF3S6, INT6 Page 1 of 2 Specificity Rabbit anti Human EIF3E antibody recognizes the eukaryotic translation initiation factor 3 subunit E, also known as eIF-3 p48, eukaryotic translation initiation factor 3 subunit 6, mammary tumor- associated protein INT6 or viral integration site protein INT-6 homolog. Rabbit anti Human EIF3E antibody detects a band of 48 kDa. -

Apoptotic Genes As Potential Markers of Metastatic Phenotype in Human Osteosarcoma Cell Lines
17-31 10/12/07 14:53 Page 17 INTERNATIONAL JOURNAL OF ONCOLOGY 32: 17-31, 2008 17 Apoptotic genes as potential markers of metastatic phenotype in human osteosarcoma cell lines CINZIA ZUCCHINI1, ANNA ROCCHI2, MARIA CRISTINA MANARA2, PAOLA DE SANCTIS1, CRISTINA CAPANNI3, MICHELE BIANCHINI1, PAOLO CARINCI1, KATIA SCOTLANDI2 and LUISA VALVASSORI1 1Dipartimento di Istologia, Embriologia e Biologia Applicata, Università di Bologna, Via Belmeloro 8, 40126 Bologna; 2Laboratorio di Ricerca Oncologica, Istituti Ortopedici Rizzoli; 3IGM-CNR, Unit of Bologna, c/o Istituti Ortopedici Rizzoli, Via di Barbiano 1/10, 40136 Bologna, Italy Received May 29, 2007; Accepted July 19, 2007 Abstract. Metastasis is the most frequent cause of death among malignant primitive bone tumor, usually developing in children patients with osteosarcoma. We have previously demonstrated and adolescents, with a high tendency to metastasize (2). in independent experiments that the forced expression of Metastases in osteosarcoma patients spread through peripheral L/B/K ALP and CD99 in U-2 OS osteosarcoma cell lines blood very early and colonize primarily the lung, and later markedly reduces the metastatic ability of these cancer cells. other skeleton districts (3). Since disseminated hidden micro- This behavior makes these cell lines a useful model to assess metastases are present in 80-90% of OS patients at the time the intersection of multiple and independent gene expression of diagnosis, the identification of markers of invasiveness signatures concerning the biological problem of dissemination. and metastasis forms a target of paramount importance in With the aim to characterize a common transcriptional profile planning the treatment of osteosarcoma lesions and enhancing reflecting the essential features of metastatic behavior, we the prognosis. -

Whole Exome Sequencing in Families at High Risk for Hodgkin Lymphoma: Identification of a Predisposing Mutation in the KDR Gene
Hodgkin Lymphoma SUPPLEMENTARY APPENDIX Whole exome sequencing in families at high risk for Hodgkin lymphoma: identification of a predisposing mutation in the KDR gene Melissa Rotunno, 1 Mary L. McMaster, 1 Joseph Boland, 2 Sara Bass, 2 Xijun Zhang, 2 Laurie Burdett, 2 Belynda Hicks, 2 Sarangan Ravichandran, 3 Brian T. Luke, 3 Meredith Yeager, 2 Laura Fontaine, 4 Paula L. Hyland, 1 Alisa M. Goldstein, 1 NCI DCEG Cancer Sequencing Working Group, NCI DCEG Cancer Genomics Research Laboratory, Stephen J. Chanock, 5 Neil E. Caporaso, 1 Margaret A. Tucker, 6 and Lynn R. Goldin 1 1Genetic Epidemiology Branch, Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD; 2Cancer Genomics Research Laboratory, Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD; 3Ad - vanced Biomedical Computing Center, Leidos Biomedical Research Inc.; Frederick National Laboratory for Cancer Research, Frederick, MD; 4Westat, Inc., Rockville MD; 5Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD; and 6Human Genetics Program, Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD, USA ©2016 Ferrata Storti Foundation. This is an open-access paper. doi:10.3324/haematol.2015.135475 Received: August 19, 2015. Accepted: January 7, 2016. Pre-published: June 13, 2016. Correspondence: [email protected] Supplemental Author Information: NCI DCEG Cancer Sequencing Working Group: Mark H. Greene, Allan Hildesheim, Nan Hu, Maria Theresa Landi, Jennifer Loud, Phuong Mai, Lisa Mirabello, Lindsay Morton, Dilys Parry, Anand Pathak, Douglas R. Stewart, Philip R. Taylor, Geoffrey S. Tobias, Xiaohong R. Yang, Guoqin Yu NCI DCEG Cancer Genomics Research Laboratory: Salma Chowdhury, Michael Cullen, Casey Dagnall, Herbert Higson, Amy A. -

Whole Genome Sequencing of Familial Non-Medullary Thyroid Cancer Identifies Germline Alterations in MAPK/ERK and PI3K/AKT Signaling Pathways
biomolecules Article Whole Genome Sequencing of Familial Non-Medullary Thyroid Cancer Identifies Germline Alterations in MAPK/ERK and PI3K/AKT Signaling Pathways Aayushi Srivastava 1,2,3,4 , Abhishek Kumar 1,5,6 , Sara Giangiobbe 1, Elena Bonora 7, Kari Hemminki 1, Asta Försti 1,2,3 and Obul Reddy Bandapalli 1,2,3,* 1 Division of Molecular Genetic Epidemiology, German Cancer Research Center (DKFZ), D-69120 Heidelberg, Germany; [email protected] (A.S.); [email protected] (A.K.); [email protected] (S.G.); [email protected] (K.H.); [email protected] (A.F.) 2 Hopp Children’s Cancer Center (KiTZ), D-69120 Heidelberg, Germany 3 Division of Pediatric Neurooncology, German Cancer Research Center (DKFZ), German Cancer Consortium (DKTK), D-69120 Heidelberg, Germany 4 Medical Faculty, Heidelberg University, D-69120 Heidelberg, Germany 5 Institute of Bioinformatics, International Technology Park, Bangalore 560066, India 6 Manipal Academy of Higher Education (MAHE), Manipal, Karnataka 576104, India 7 S.Orsola-Malphigi Hospital, Unit of Medical Genetics, 40138 Bologna, Italy; [email protected] * Correspondence: [email protected]; Tel.: +49-6221-42-1709 Received: 29 August 2019; Accepted: 10 October 2019; Published: 13 October 2019 Abstract: Evidence of familial inheritance in non-medullary thyroid cancer (NMTC) has accumulated over the last few decades. However, known variants account for a very small percentage of the genetic burden. Here, we focused on the identification of common pathways and networks enriched in NMTC families to better understand its pathogenesis with the final aim of identifying one novel high/moderate-penetrance germline predisposition variant segregating with the disease in each studied family. -

Open Data for Differential Network Analysis in Glioma
International Journal of Molecular Sciences Article Open Data for Differential Network Analysis in Glioma , Claire Jean-Quartier * y , Fleur Jeanquartier y and Andreas Holzinger Holzinger Group HCI-KDD, Institute for Medical Informatics, Statistics and Documentation, Medical University Graz, Auenbruggerplatz 2/V, 8036 Graz, Austria; [email protected] (F.J.); [email protected] (A.H.) * Correspondence: [email protected] These authors contributed equally to this work. y Received: 27 October 2019; Accepted: 3 January 2020; Published: 15 January 2020 Abstract: The complexity of cancer diseases demands bioinformatic techniques and translational research based on big data and personalized medicine. Open data enables researchers to accelerate cancer studies, save resources and foster collaboration. Several tools and programming approaches are available for analyzing data, including annotation, clustering, comparison and extrapolation, merging, enrichment, functional association and statistics. We exploit openly available data via cancer gene expression analysis, we apply refinement as well as enrichment analysis via gene ontology and conclude with graph-based visualization of involved protein interaction networks as a basis for signaling. The different databases allowed for the construction of huge networks or specified ones consisting of high-confidence interactions only. Several genes associated to glioma were isolated via a network analysis from top hub nodes as well as from an outlier analysis. The latter approach highlights a mitogen-activated protein kinase next to a member of histondeacetylases and a protein phosphatase as genes uncommonly associated with glioma. Cluster analysis from top hub nodes lists several identified glioma-associated gene products to function within protein complexes, including epidermal growth factors as well as cell cycle proteins or RAS proto-oncogenes. -

Integrated Data Analysis Reveals Uterine Leiomyoma Subtypes with Distinct Driver Pathways and Biomarkers
Integrated data analysis reveals uterine leiomyoma subtypes with distinct driver pathways and biomarkers Miika Mehinea,b, Eevi Kaasinena,b, Hanna-Riikka Heinonena,b, Netta Mäkinena,b, Kati Kämpjärvia,b, Nanna Sarvilinnab,c, Mervi Aavikkoa,b, Anna Vähärautiob, Annukka Pasanend, Ralf Bützowd, Oskari Heikinheimoc, Jari Sjöbergc, Esa Pitkänena,b, Pia Vahteristoa,b, and Lauri A. Aaltonena,b,e,1 aMedicum, Department of Medical and Clinical Genetics, University of Helsinki, Helsinki FIN-00014, Finland; bResearch Programs Unit, Genome-Scale Biology, University of Helsinki, Helsinki FIN-00014, Finland; cDepartment of Obstetrics and Gynecology, Helsinki University Hospital, University of Helsinki, Helsinki FIN-00029, Finland; dDepartment of Pathology and HUSLAB, Helsinki University Hospital, University of Helsinki, Helsinki FIN-00014, Finland; and eDepartment of Biosciences and Nutrition, Karolinska Institutet, SE-171 77, Stockholm, Sweden Edited by Bert Vogelstein, Johns Hopkins University, Baltimore, MD, and approved December 18, 2015 (received for review September 25, 2015) Uterine leiomyomas are common benign smooth muscle tumors that with deletions affecting collagen, type IV, alpha 5 and collagen, type impose a major burden on women’s health. Recent sequencing studies IV, alpha 6 (COL4A5-COL4A6) may constitute a rare fourth subtype have revealed recurrent and mutually exclusive mutations in leiomyo- (4). HMGA2 and MED12 represent the two most common driver mas, suggesting the involvement of molecularly distinct pathways. In genes and together contribute to 80–90% of all leiomyomas (5). this study, we explored transcriptional differences among leiomyomas Less frequently, leiomyomas harbor 6p21 rearrangements af- harboring different genetic drivers, including high mobility group fecting high mobility group AT-hook 1 (HMGA1) (6). -
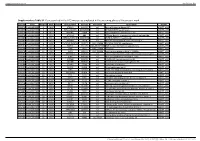
Supplementary Table S1. Genes Printed in the HC5 Microarray Employed in the Screening Phase of the Present Work
Supplementary material Ann Rheum Dis Supplementary Table S1. Genes printed in the HC5 microarray employed in the screening phase of the present work. CloneID Plate Position Well Length GeneSymbol GeneID Accession Description Vector 692672 HsxXG013989 2 B01 STK32A 202374 null serine/threonine kinase 32A pANT7_cGST 692675 HsxXG013989 3 C01 RPS10-NUDT3 100529239 null RPS10-NUDT3 readthrough pANT7_cGST 692678 HsxXG013989 4 D01 SPATA6L 55064 null spermatogenesis associated 6-like pANT7_cGST 692679 HsxXG013989 5 E01 ATP1A4 480 null ATPase, Na+/K+ transporting, alpha 4 polypeptide pANT7_cGST 692689 HsxXG013989 6 F01 ZNF816-ZNF321P 100529240 null ZNF816-ZNF321P readthrough pANT7_cGST 692691 HsxXG013989 7 G01 NKAIN1 79570 null Na+/K+ transporting ATPase interacting 1 pANT7_cGST 693155 HsxXG013989 8 H01 TNFSF12-TNFSF13 407977 NM_172089 TNFSF12-TNFSF13 readthrough pANT7_cGST 693161 HsxXG013989 9 A02 RAB12 201475 NM_001025300 RAB12, member RAS oncogene family pANT7_cGST 693169 HsxXG013989 10 B02 SYN1 6853 NM_133499 synapsin I pANT7_cGST 693176 HsxXG013989 11 C02 GJD3 125111 NM_152219 gap junction protein, delta 3, 31.9kDa pANT7_cGST 693181 HsxXG013989 12 D02 CHCHD10 400916 null coiled-coil-helix-coiled-coil-helix domain containing 10 pANT7_cGST 693184 HsxXG013989 13 E02 IDNK 414328 null idnK, gluconokinase homolog (E. coli) pANT7_cGST 693187 HsxXG013989 14 F02 LYPD6B 130576 null LY6/PLAUR domain containing 6B pANT7_cGST 693189 HsxXG013989 15 G02 C8orf86 389649 null chromosome 8 open reading frame 86 pANT7_cGST 693194 HsxXG013989 16 H02 CENPQ 55166 -

EIF3E (NM 001568) Human Recombinant Protein Product Data
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for TP304788 EIF3E (NM_001568) Human Recombinant Protein Product data: Product Type: Recombinant Proteins Description: Recombinant protein of human eukaryotic translation initiation factor 3, subunit E (EIF3E) Species: Human Expression Host: HEK293T Tag: C-Myc/DDK Predicted MW: 52 kDa Concentration: >50 ug/mL as determined by microplate BCA method Purity: > 80% as determined by SDS-PAGE and Coomassie blue staining Buffer: 25 mM Tris.HCl, pH 7.3, 100 mM glycine, 10% glycerol Preparation: Recombinant protein was captured through anti-DDK affinity column followed by conventional chromatography steps. Storage: Store at -80°C. Stability: Stable for 12 months from the date of receipt of the product under proper storage and handling conditions. Avoid repeated freeze-thaw cycles. RefSeq: NP_001559 Locus ID: 3646 UniProt ID: P60228 RefSeq Size: 1516 Cytogenetics: 8q23.1 RefSeq ORF: 1335 Synonyms: eIF3-p46; EIF3-P48; EIF3S6; INT6 This product is to be used for laboratory only. Not for diagnostic or therapeutic use. View online » ©2021 OriGene Technologies, Inc., 9620 Medical Center Drive, Ste 200, Rockville, MD 20850, US 1 / 2 EIF3E (NM_001568) Human Recombinant Protein – TP304788 Summary: Component of the eukaryotic translation initiation factor 3 (eIF-3) complex, which is required for several steps in the initiation of protein synthesis (PubMed:17581632, PubMed:25849773, PubMed:27462815). The eIF-3 complex associates with the 40S ribosome and facilitates the recruitment of eIF-1, eIF-1A, eIF-2:GTP:methionyl-tRNAi and eIF-5 to form the 43S pre- initiation complex (43S PIC). -

Genename Sequence Peptidecount
GeneName Sequence PeptideCount A1BG SLPAPWLSMAPVSWITPGLK 1 A1S9T;UBA1;UBE1 AAVATFLQSVQVPEFTPK 18 A1S9T;UBA1;UBE1 AENYDIPSADR 18 A1S9T;UBA1;UBE1 ALPAVQQNNLDEDLIR 18 A1S9T;UBA1;UBE1 ALPAVQQNNLDEDLIRK 18 A1S9T;UBA1;UBE1 ATLPSPDKLPGFK 18 A1S9T;UBA1;UBE1 DEFEGLFK 18 A1S9T;UBA1;UBE1 GGIVSQVK 18 A1S9T;UBA1;UBE1 GLGVEIAK 18 A1S9T;UBA1;UBE1 HQYYNQEWTLWDR 18 A1S9T;UBA1;UBE1 IYDDDFFQNLDGVANALDNVDAR 18 A1S9T;UBA1;UBE1 KPLLESGTLGTK 18 A1S9T;UBA1;UBE1 LAGTQPLEVLEAVQR 18 A1S9T;UBA1;UBE1 LQTSSVLVSGLR 18 A1S9T;UBA1;UBE1 NEEDAAELVALAQAVNAR 18 A1S9T;UBA1;UBE1 NFPNAIEHTLQWAR 18 A1S9T;UBA1;UBE1 QFLDYFK 18 A1S9T;UBA1;UBE1 QFLFRPWDVTK 18 A1S9T;UBA1;UBE1 SLVASLAEPDFVVTDFAK 18 A2LD1 TLEPYPLVIAGEHNIPWLLHLPGSGR 1 A4;AD1;APP THPHFVIPYR 2 A4;AD1;APP WYFDVTEGK 2 AAAS;ADRACALA;GL003 GGGVTNLLWSPDGSK 12 AAAS;ADRACALA;GL003 GQWINLPVLQLTK 12 AAAS;ADRACALA;GL003 IAHIPLYFVNAQFPR 12 AAAS;ADRACALA;GL003 ILATTPSAVFR 12 AAAS;ADRACALA;GL003 LAVLMK 12 AAAS;ADRACALA;GL003 LLSASPVDAAIR 12 AAAS;ADRACALA;GL003 SATIVADLSETTIQTPDGEER 12 AAAS;ADRACALA;GL003 VFAWHPHTNK 12 AAAS;ADRACALA;GL003 VQDGKPVILLFR 12 AAAS;ADRACALA;GL003 VWEAQMWTCER 12 AAAS;ADRACALA;GL003 VYNASSTIVPSLK 12 AAAS;ADRACALA;GL003 WPTLSGR 12 AAC4;ANT4;SFEC;SLC25A31 DLLAGGVAAAVSK 21 AAC4;ANT4;SFEC;SLC25A31 EQGFFSFWR 21 AAC4;ANT4;SFEC;SLC25A31 GAFSNVLR 21 AAC4;ANT4;SFEC;SLC25A31 GLGDCIMK 21 AAC4;ANT4;SFEC;SLC25A31 GLLPKPK 21 AAC4;ANT4;SFEC;SLC25A31 GMVDCLVR 21 AAC4;ANT4;SFEC;SLC25A31 GNLANVIR 21 AAC4;ANT4;SFEC;SLC25A31 GTGGALVLVLYDK 21 AAC4;ANT4;SFEC;SLC25A31 IKEFFHIDIGGR 21 AAC4;ANT4;SFEC;SLC25A31 IPREQGFFSFWR 21 AAC4;ANT4;SFEC;SLC25A31